Protein Folding in Human Health: 2019 Dr. Paul Janssen Award Symposium
Reported by:
Diana Gitig
Presented by:
The Dr. Paul Janssen Award for Biomedical Research
The New York Academy of Sciences
Overview
Chaperones bind to peptide chains as they are being transcribed to prevent them from aggregating and to give them an isolated, quiet space, shielded from the hubbub of the crowded cytoplasm, in which to fold properly. This process is essential to human biology and health, because misfolded proteins are associated with aging and diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and prion disease.
On October 4, 2019, prominent scientists gathered at the New York Academy of Sciences to grant the 2019 Dr. Paul Janssen Award to Hartl and Horwich for their groundbreaking insights into chaperone-mediated protein folding. The symposium included award lectures from the honorees, as well as presentations on several aspects of protein folding, from basic biology to the implications for human disease.
Symposium Highlights
- While studying mitochondrial protein import, Horwich and Hartl hypothesized that the process may not be spontaneous but dependent on cellular machinery. They discovered a new class of proteins responsible for protein folding.
- Hsp60, its bacterial homolog GroEL, and its eukaryotic homolog TRiC have a double ring structure that forms a chamber in which a peptide substrate can fold into its proper shape.
- The unfolded protein response of the endoplasmic reticulum responds to the presence of misfolded proteins, which accrue with age. The response itself declines with age.
- Hsp70 is a diverse family of monomeric chaperones that binds to polypeptide chains as they’re being translated or when they misfold from mutation or stress and prevents them from collapsing into aggregates.
- Clinically relevant receptors that have been difficult to treat require specific chaperones that may provide more easily druggable targets for neurological and psychiatric disorders.
Honorees
Franz-Ulrich Hartl, MD
Max Planck Institute of Biochemistry
Arthur Horwich, MD
Yale School of Medicine and Howard Hughes Medical Institute
Speakers
David S. Bredt, MD, PhD
Janssen Pharmaceutical Companies of Johnson & Johnson
Andrew Dillin, PhD
University of California, Berkeley and Howard Hughes Medical Institute
Judith Frydman, PhD
Stanford University
Lila M. Gierasch, PhD
University of Massachusetts Amherst
Event Sponsors
This symposium was made possible with support from:

Dr. Paul Janssen Award Lectures
Speakers
Franz-Ulrich Hartl
Max Planck Institute of Biochemistry
Arthur Horwich
Yale School of Medicine and Howard Hughes Medical Institute
Highlights
- Chaperones prevent the formation of toxic protein aggregates, and failure of the chaperone system is associated with numerous age-dependent proteopathies and neurodegenerative diseases.
- GroEL mediates two key actions on a substrate polypeptide: binding in the open ring forestalls aggregation and can exert unfolding, while binding in the closed ring holds the polypeptide in “solitary confinement,” giving it a chance to fold on its own and alleviating the risk of aggregation.
Molecular Chaperones — Central Players of the Proteostasis Network
“Protein folding is the final step in the information transfer from gene to functional protein, and as such is of fundamental biological importance,” began Franz-Ulrich Hartl.
In the 1950s, biochemist Christian Anfinsen showed that denatured proteins could refold spontaneously in vitro, thus revealing that all of the information required for a protein to attain its final structure is contained in its amino acid sequence. The study was somewhat misleading, however, as it only used small proteins — under 100 amino acids long — and it started with a completely synthesized amino acid chain. This hardly recapitulates the conditions under which proteins must fold in the cell, where many proteins are large, have multiple domains, fold as they are being synthesized on the ribosome, and are in the very crowded cytoplasm.
In the late 1980s, growing evidence showed that cellular machines were required to help proteins fold “at biologically relevant timescales.” These machines were deemed molecular chaperones, as they help proteins achieve their final active conformations but are not themselves part of the final structure. Hartl and Horwich initially discovered chaperones using mitochondria as a model system.
Mitochondria import about 1,000 proteins from the cytoplasm, and these proteins must be unfolded to get across the mitochondrial membranes. Based on Anfinsen’s experiments, it was thought that they would then spontaneously fold properly once inside the mitochondria. But proteins in yeast with mutant Hsp60 got into the mitochondria but failed to fold, identifying Hsp60 as a required chaperone.
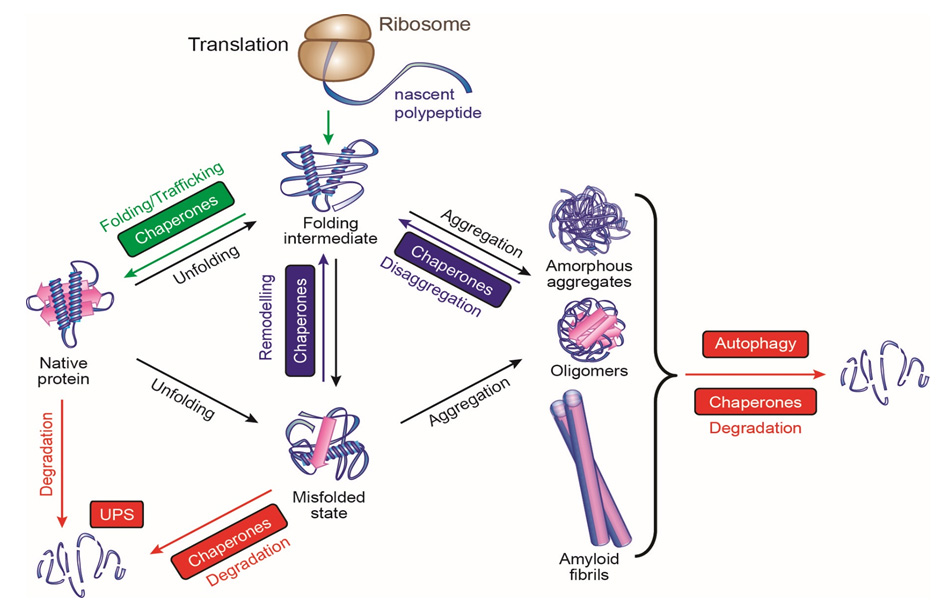
Chaperones like Hsp60 prevent the formation of protein aggregates. Aggregation can occur in the intermediate stages of multidomain protein folding when hydrophobic regions might become exposed; chaperones protect these hydrophobic regions through multiple rounds of binding and releasing the partially folded proteins.
ATP binding and hydrolysis often mediate these bind-and-release cycles. The chaperones provide a safe space for the proteins to fold, sequestered away from the hubbub of the cytoplasm. Proteins revisit the quiet chambers that chaperones provide throughout their lifetimes, not only as they are being synthesized.
In the current model, while an amino acid chain is being translated, it interacts with a nascent-chain-binding protein like Hsp70, a type of chaperone that binds to hydrophobic peptide segments. Hsp70 prevents premature misfolding, only allowing the protein to fold when enough structural information for productive folding becomes available — when the protein chain gets long enough.
Most proteins only require this type of chaperone to fold efficiently. But some have more complicated structures and need to fold in the isolated, constrained cage of a cylindrical chaperonin complex like Hsp60, the chaperone that Hartl and Horwich first isolated from mitochondria. Bacterial GroEL and its cofactor GroES are the most well-studied of this class of chaperones; the eukaryotic cytoplasmic versions are called TRiC or CCT.
There is an age-dependent decline in chaperone function, though. Since chaperones are required for protein maintenance, this decline can lead to a buildup of protein aggregates — which then further strains the already declining chaperones.
These protein aggregates lead to neurodegenerative diseases like Alzheimer’s disease and Huntington’s disease. Aggregates of different disease proteins have the same amyloid fibrillar structure, which suggests that a basic pathological mechanism may underlie all of these diseases. Hartl found that the aggregates interfere with almost every aspect of cellular machinery — transcription, translation, nuclear translocation, DNA maintenance, protein degradation, cytoskeletal organization, and vesicle transport —not only chaperones. But as they overwhelm the chaperone system, toxic aggregates build up until they cause cell death.
Thus, he suggests that rebalancing the proteostasis network may be a means of treating these neurodegenerative diseases.
Chaperonin-mediated Protein Folding
Arthur Horwich described how, in a classic bedside-to-bench approach, he discovered that chaperonin ring machines function to mediate protein folding. He studied the lethal X linked inherited metabolic disease caused by the mutant mitochondrial enzyme OTC. OTC is the second step in the urea cycle; when it is defective, cells can’t clear urea.
Since it is X linked, baby boys with nonfunctional OTC die. Horwich isolated the OTC cDNA and found its mitochondrial transport signal, then looked for a yeast mutant that could transport unfolded human OTC into the mitochondria but in which the transported OTC would not then fold. The yeast mutant he found lacked Hsp60.
Mitochondrial Hsp60, and its bacterial counterpart GroEL, performs two vital functions: they bind to polypeptides to prevent the formation of protein aggregates, and they help polypeptides achieve their functional state. In 1994 and 1997, the X-ray structures of both GroEL alone and in complex with its cochaperonin single ring GroES were presented along with structure-function studies in collaborative work with the late Paul Sigler, providing insight into how the machinery works.
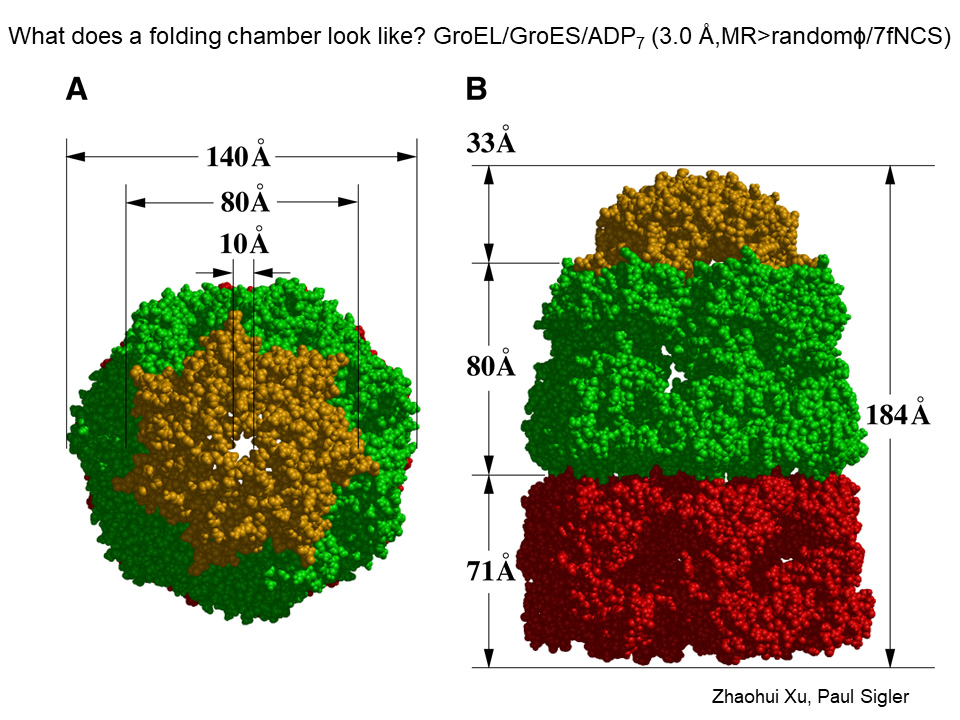
GroEL is a cylinder made of 14 identical subunits arranged into two back-to-back 7-membered rings. Each of the subunits is folded into: an equatorial domain, at the waistline of the cylinder, the collective of which hold the assembly together via side-by-side contacts within a ring and contacts of subunits between the two rings; a hinge like “intermediate” domain interconnecting the equatorial and apical domain; and a terminal “apical” domain at an end of the cylinder.
The equatorial domains each house an ATP binding pocket at the inside aspect and the cooperative binding of 7 ATP’s in a GroEL ring causes the terminal GroEL apical domains, attached to the equatorial domains through the slender intermediate domains, to open up like flower petals. In their “unopened” position the apical domains surround an open central cavity of 45 Angstrom diameter and each apical domain proffers sticky “hydrophobic” surface at its cavity-facing aspect.
The continuous hydrophobic surface around the ring specifically captures an unfolded protein species via its own exposed hydrophobic surface (that will become buried to the interior in the final folded “native” form). Thus the binding of a non-native protein by an open GroEL ring serves to capture the protein’s sticky hydrophobic surfaces, masking them, and preventing them from interacting with other unfolded proteins which can lead to aggregation.
When a polypeptide-bound ring of GroEL binds the cochaperonin ring, GroES, a smaller 7-membered single ring of identical subunits, in the presence of ATP, now a large movement of the apical domains occurs, both clockwise rotation and further elevation (see Figure; GroES is colored gold and the GroEL ring undergoing large movements is green). The large movements remove the hydrophobic polypeptide binding surface from facing the cavity, and the lining of the now GroES-encapsulated GroEL cavity becomes watery (hydrophilic) in character.
The large twisting apical domain movements strip the polypeptide off of the cavity wall into the now encapsulated and watery (hydrophilic) cavity where the protein folds in “solitary confinement,” as Horwich phrased it, without any chance of aggregation. Subsequently, after this longest step of the reaction cycle (~10 sec), ATP hydrolyzes, GroES releases, and out from the cavity comes the polypeptide whether properly folded or not. If it has not reached native form, it can make another try at proper folding, either by entering another GroEL cavity, or becoming bound to a different chaperone.
Further Readings
Hartl
Balchin D, Hayer-Hartl M, et al.
In vivo aspects of protein folding and quality control.
Science. 2016 Jul 1;353(6294).
Frydman J, Nimmesgern E, Ohtsuka K, et al.
Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones.
Nature. 1994 Jul 14;370(6485):111-7.
Hipp MS, Park SH, Hartl FU.
Proteostasis impairment in protein-misfolding and -aggregation diseases.
Trends Cell Biol. 2014 Sep;24(9):506-14.
Horwich
Elad N, Farr GW, Clare DK, et al.
Topologies of a substrate protein bound to the chaperonin GroEL.
Mol Cell. 2007 May 11;26(3):415-26.
Weissman JS, Hohl CM, Kovalenko O, et al.
Cell. 1995 Nov 17;83(4):577-87.
Xu Z, Horwich AL, Sigler PB.
The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex.
Nature. 1997 Aug 21;388(6644):741-50.
Advances in Protein Folding
Speakers
Judith Frydman
Stanford University
Andrew Dillin
University of California, Berkeley and Howard Hughes Medical Institute
Highlights
- There are a considerable variety of chaperones that are structurally and functionally different from recognizing and binding nonnative proteins in all of their various stages and processes.
- The endoplasmic reticulum unfolded protein response evolved to protect the organism from infection. In the nervous system, it can act in a non-autonomous manner to promote transcription in response to stress.
The TRiCKy Business of Folding Proteins in the Cell
“Proteins are astoundingly complex,” said Judith Frydman. As an example, she pointed to the mammalian respiratory complex I, the 45-subunit complex that drives protons across the inner mitochondrial membrane. Thus, the potential problems with protein folding are not limited to the folding process.
Chaperones bind unfolded polypeptides to help them achieve their native state. Still, much more than that, they engage polypeptides at every stage of their existence in the cell, waiting to receive them as they’re translated and monitoring for damage throughout their lifespans.
TRiC, or CCT, is the stacked chaperone in eukaryotic cells — the equivalent of GroEL. However, unlike GroEL, it does not have a separate cap. It requires ATP hydrolysis, which closes the lid to allow folding; but ATP binding is not sufficient. TRiC binds nascent chains when they are almost complete, while they are still on the ribosome but after they have interacted with Hsp70.
The complex only binds precise types of folding intermediates — notably those with complex topologies like p53, tubulin, actin, telomerase, F box proteins, and others — and then comes off once that folding intermediate has resolved into its properly folded domain. It also suppresses amyloid aggregation, but is overexpressed in many cancers and has been linked to poor prognosis in lung and breast cancer.
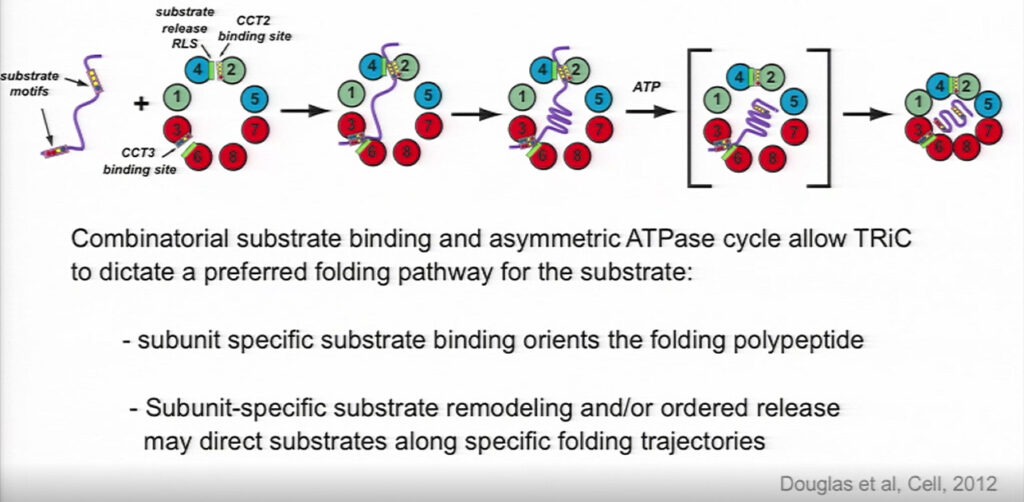
TRiC descends from the chaperone in archaea, which only has one type of subunit. The heteromeric nature of eukaryotic TRiC allows it to form an asymmetrical complex. TRiC has eight subunits, and each subunit has a different affinity for ATP; these subunits are arranged with high-affinity subunits around one side of the ring and low-affinity subunits around the other side.
The subunits have varying degrees of affinity for substrates as well, with each subunit’s binding site presenting a distinct and evolutionarily conserved surface of polar and hydrophobic residues. Their combination thus broadens TRiC’s binding specificity.
Once the binding chamber is closed, one hemisphere is positively charged and the other is negatively charged, further orienting how the substrate can bind and influencing its folding trajectory. Frydman called it a “chaperone with an opinion,” rather than a cage, “that guides the substrate where it needs to go.”
Prefoldin is a cofactor for TRiC, so named because it was thought to facilitate substrate transfer to TRiC before the substrate folded. It binds to TRiC in TRiC’s open state, and, like TRiC, it has a charge asymmetry and a specific pattern of polar and hydrophobic residues that contribute to the inner surface of TRiC’s binding chamber. Prefoldin seems to enhance both the yield and the rate of folding. In vivo, it must bind to TRiC, or else massive protein aggregation builds up in the cell.
Perceiving ER Stress
As many as thirteen million proteins fold and mature in the endoplasmic reticulum (ER) every minute. It is no wonder then that defects in ER function are strongly associated with metabolic and age-related disorders. The unfolded protein response in the ER (UPRER) responds to the presence of unfolded proteins by inducing the transcription of chaperones, and it declines with age. Andrew Dillin wondered how this UPRER works in multicellular organisms.
Are unfolded proteins detected in each individual cell by its own machinery, in a stochastic manner? Or might there be a higher order of regulation, coordinating protein folding mechanisms across the whole system? He turned to C. elegans to figure it out. Since all of the cells in the adult C. elegans are post mitotic, the worm provides a great model system for studying proteome maintenance.
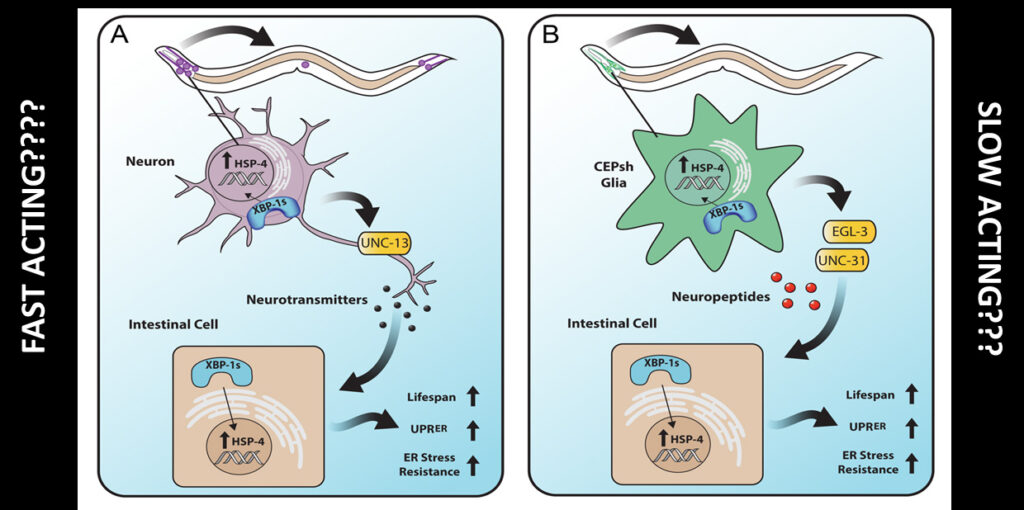
The Dillin lab demonstrated that the neuronal transcription factor XBP-1 could rescue the age-dependent decline in ER proteostasis. Overexpression of XBP-1 extends the worm’s life. XBP-1 — which has the very unusual property that its mRNA is spliced in the cytoplasm instead of the nucleus — senses unfolded proteins and induces the UPRER in nerve cells. These nerves then send signals to peripheral and distal cells, causing them to activate their own UPRER.
Only neuronal cells, both neurons and glia, respond to XBP by inducing the UPR. The peripheral cells don’t sense the unfolded proteins and respond to them; they respond to the signal from the brain. Neurons require small, clear vesicles to send this signal, indicating that neurotransmitters are involved. Unlike neurons, glia need dense core vesicles, suggesting that they signal through neuropeptides or biologic amines rather than neurotransmitters. The neuronal and glial effects are synergistic, and the mechanism is conserved in mice.
The UPRER “only deals with the challenge after the damage has occurred” said Dillin. Wouldn’t a protective system be preferable?
Thus, he conducted a CRISPR screen to find such a system, of UPRER regulators that would identify and protect the organism from ER stress instead of just responding after it happens. In doing so, Dillin found TMEM2, a transmembrane hyaluronidase that had not been previously implicated in ER stress. It does not activate the UPRER, which can induce apoptosis. Rather, it acts through the MAP kinase pathway to promote stress resistance in the ER and survival of the organism.
By breaking down extracellular hyaluronan, it generates a smaller product that increases ER stress resistance. TMEM2 is conserved from worms all the way through humans; it senses the stress from outside the plasma membrane of brain cells, before the stress hits, and then sends the signal to the periphery. Dillin does not yet know how TMEM protects the ER from stress, but he knows that it is not through chaperones.
Further Readings
Frydman
Gestaut D, Limatola A, Joachimiak L, et al.
Curr Opin Struct Biol. 2019 Apr;55:50-58.
Gestaut D, Roh SH, Ma B, et al.
Cell. 2019 Apr 18;177(3):751-765.e15.
Dillin
Frakes AE, Dillin A.
The UPRER: Sensor and Coordinator of Organismal Homeostasis.
Mol Cell. 2017 Jun 15;66(6):761-771.
Protein Folding and Drug Development
Speakers and Panelists
Franz-Ulrich Hartl
Max Planck Institute of Biochemistry
Arthur Horwich
Yale School of Medicine and Howard Hughes Medical Institute
Lila M. Gierasch
University of Massachusetts Amherst
David S. Bredt
Janssen Pharmaceutical Companies of Johnson & Johnson
Seema Kumar (Moderator)
Johnson & Johnson
Highlights
- The Hsp70 allosteric cycle involves major conformational changes, alternating between a docked state with bound ATP and low affinity for unfolded protein substrates and an undocked state in which the α-helical lid rotates out of the way to allow substrate binding and ATP hydrolysis.
- Receptors implicated in neuronal and psychiatric disorders often require specific chaperones to help them fold; these chaperones are often expressed only in specific areas of the brain, and thus may provide appropriate drug targets.
The Versatile Hsp70 Molecular Chaperones Machine
Lila Gierasch introduced Hsp70 as the “early greeting committee” for nascent polypeptide chains. It can maintain the chains in an unfolded state for transport across membranes and meet them on the other side. Hsp70 can also give them a second chance to fold if things don’t go right the first time around. Like all chaperones, it prevents aggregation. It acts as a monomer, but that hardly makes it simple.
Hsp70 activities depend on intramolecular allostery controlled by ligand modulation of an energy landscape. The C-terminal substrate-binding domain (SBD) binds to short hydrophobic stretches of a polypeptide chain. ATP binding to the N-terminal nucleotide-binding domain (NBD) reorients the NBD actin fold. It decreases the affinity of the SBD for the substrate, and the substrate activates the NBD ATPase activity. The α-helical lid can rotate, allowing access to either the SBD or the NBD.
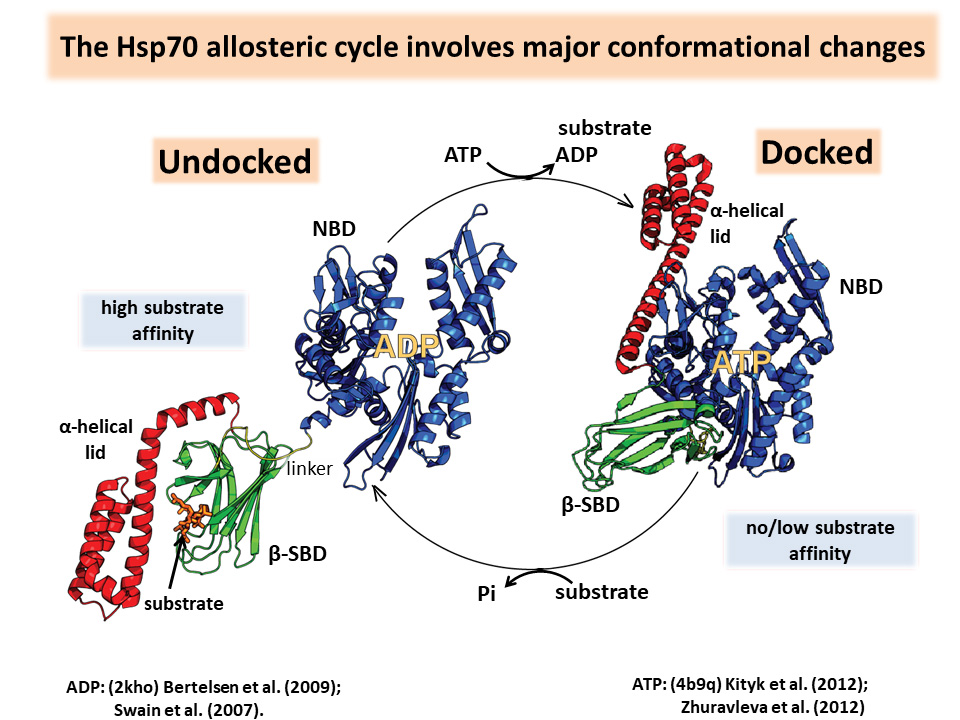
Hsp70 allosteric landscapes can be shaped by the strength of interdomain interfaces and as well as ligand binding, making them “tunable molecular machines.” They must have promiscuous selectivity because they bind an immense number of substrates with varying affinities.
There are Hsp70 molecules bound approximately every 40 amino acids throughout the proteome, and there is evidence that more than one Hsp70 molecule can bind to one substrate, mainly to keep it unfolded as it is translocated. And there are many isoforms of eukaryotic Hsp70 with different allosteries. These could have evolved through interactions with co-chaperones, post-translational modifications like phosphorylation, and even the sequence of the substrate.
Gierasch suggested that tweaking its allostery might modulate Hsp70 activity, or one class of Hsp70 could be targeted over another to treat particular diseases. It is tempting to think of activating the chaperone network to prevent neurodegeneration, but it is risky, too, since cancer cells often rely on mutant chaperones.
Getting a Handle on Neuropharmacology by Targeting Receptor Chaperones
Abnormalities in psychiatric diseases are heterogeneous across brain regions, with increased activity in some areas and decreased activity in others. It has been very difficult to find small molecules that can affect synaptic transmission in these different regions.
Stargazer mutant mice, that constantly look up because they have epilepsy, don’t have functional AMPARs (a type of glutamate receptor) on their cerebellar granule cells. David Brendt found that the receptors didn’t work because the mice lacked a chaperone he named stargazin. Stargazin is a Transmembrane AMPAR Regulatory Protein, or TARP, a family of proteins that Bredt said, “act more like escorts than chaperones.”
TARPs take the AMPARs from the endoplasmic reticulum to the cell surface at the synapse of cerebellar granule cells. Different TARPs are distributed to different brain regions, making them attractive drug targets. A molecule that disrupts the interaction between TARP-γ8 and AMPAR has been shown to inhibit neurotransmission in the hippocampus.
Thus, TARPs could be key to treating epilepsy without the terrible side effects of current anticonvulsants, and could possibly be used to treat bipolar disorder, schizophrenia, and anxiety.
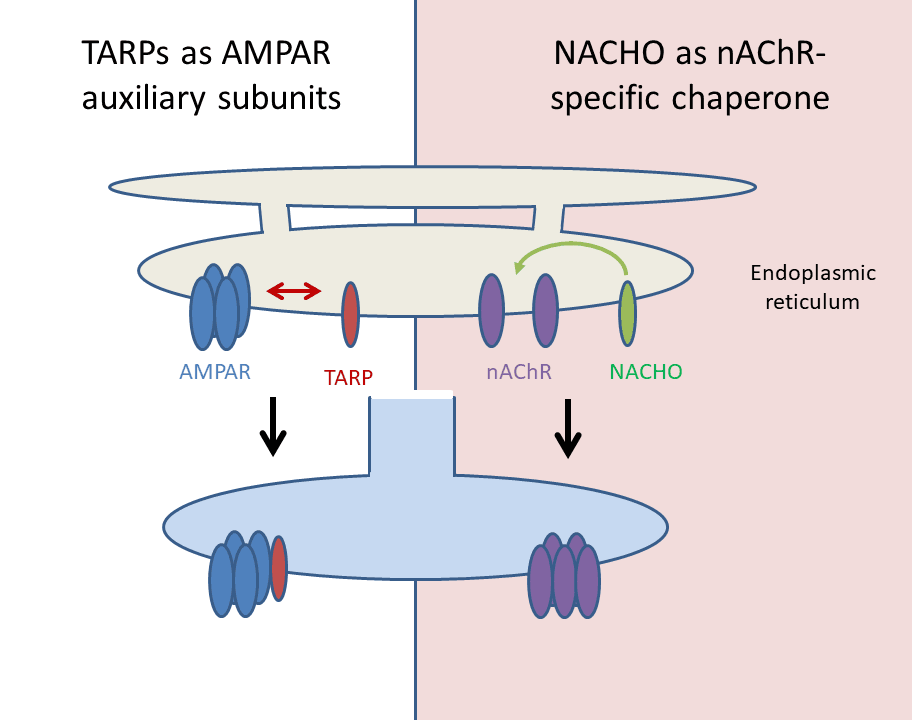
Acetylcholine receptors are the site of action for a number of Alzheimer’s drugs that induce modest but reproducible improvements in cognition. These pentameric receptors have been very difficult to study in the lab, though, because they only fold properly in neuronal cells.
Bredt recognized this as an opportunity in addition to a challenge. His lab cotransfected a library of 4,000 transmembrane proteins along with the acetylcholine receptor into HEK cells and screened for any that would help the receptors fold. Only one did, a novel transmembrane protein with no homology to anything, found in one copy in mammals and Drosophila and not found in worms or yeast at all. They named it NACHO. It resides in the membrane of the endoplasmic reticulum in neuronal cells, and it mediates the folding of nicotinic acetylcholine receptors.
Panel Discussion
Highlights
- We don’t know why protein aggregates are toxic, or why chaperones’ ability to prevent their formation wanes with age.
- Future research should focus on understanding the proteostasis network in a physiological context and figuring out if, and how, it is an appropriate clinical target.
The day ended with a panel discussion in which Hartl and Horwich fielded questions. Many of them focused on the role misfolded proteins play in disease, why they accumulate with age, and if, when, and how the proteostasis machinery can be targeted therapeutically.
Moderator Seema Kumar began the panel by asking about the greatest challenges and limitations in the field. Horwich replied that we don’t understand the toxicity of misfolded proteins; we don’t even know if they themselves are toxic, or if they are recruiting other toxic mediators. He speculated that it would be great if we could monitor single polypeptide chains as they fold, to see which ones go astray and how that makes them toxic.
Since antibodies against amyloid plaques have been ineffective in Alzheimer’s disease, enhancing multiple parts of the proteostasis network might be a better strategy than targeting specific misfolded proteins or chaperones. Horwich also pointed out that we don’t know why aging thwarts chaperones: does their ability to handle their task decline, or are there genomic or proteomic issues? Hartl added that we don’t understand neurodegenerative diseases nearly well enough to know the role that protein folding plays in their development; Parkinson’s disease, for instance, is likely more than one monolithic disease.
As for how the field will unfold in the future, Horwich noted that most of what we know about protein folding mechanisms comes from in vitro studies with purified components. So we need to know more about how the cellular milieu affects binding affinities and folding. It would be helpful to determine how many times a particular ligand comes back to a particular chaperone. Hartl explained the importance of figuring out who the first responders are, who the next responders are, and if we can develop small molecules to affect the proteostasis machinery.
Further Readings
Gierasch
Zhuravleva A, Clerico EM, Gierasch LM.
Cell. 2012 Dec 7;151(6):1296-307.
Bredt
Chen L, Chetkovich DM, Petralia RS, et al.
Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms.
Nature. 2000 Dec 21-28;408(6815):936-43.
Matta JA, Gu S, Davini WB, et al.
NACHO Mediates Nicotinic Acetylcholine Receptor Function throughout the Brain.
Cell Rep. 2017 Apr 25;19(4):688-696. doi: 10.1016/j.celrep.2017.04.008.