Transformative Research in Rare Diseases: 2020 Innovators in Science Award Symposium
Reported by:
Alan Dove
Presented by:
Takeda Pharmaceutical Company Limited
The New York Academy of Sciences
By definition, a rare disease is one that afflicts relatively few people compared to the general population. Collectively, though, there are over 7,000 of these conditions known, causing immense suffering for an estimated 300 million patients. Because most rare diseases stem from specific genetic mutations, they’ve proven difficult to treat.
Genome sequencing and molecular medicine might soon change those grim statistics, though. For example, using short DNA or RNA sequences complementary to the messenger RNA for a gene, researchers can inhibit the expression of the associated protein. These complementary sequences—called antisense oligonucleotides—could soon be delivered as drugs to treat many rare diseases.
On October 2, 2020, The New York Academy of Sciences and Takeda Pharmaceuticals hosted the Frontiers in Rare Diseases: 2020 Innovators in Science Award Symposium, an event highlighting breakthroughs in rare diseases research and honoring 2020 Innovators in Science Award Winners Adrian Krainer, PhD and Jeong Ho Lee, MD, PhD. Presentations, a panel discussion, and a virtual poster session covered the basic science, recent clinical breakthroughs, and remaining challenges in this rapidly evolving field.
Symposium Highlights
- While many rare diseases are inherited, others arise through mutations in somatic cells during life.
- Antisense oligonucleotides can alter the expression of specific genes, potentially mitigating or reversing many genetic diseases.
- Clinical trials for rare disease therapies must be tailored to the pathogenesis of each disease.
- Redirecting neural stem cells to become neurons could treat many neurodegenerative diseases.
- The COVID-19 pandemic is inspiring new collaborations that could be adapted to rare disease research.
Speakers
Jeong Ho Lee, MD, PhD
Korea Advanced Institute of Science and Technology
Adrian Krainer, PhD
Cold Spring Harbor Laboratory
Annemieke Aartsma-Rus, PhD
Leiden University Medical Center
Don Cleveland, MD, PhD
University of California, San Diego
Huda Zoghbi, MD
Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital
Brad Margus
Cerevance
Graciana Diez-Roux, PhD
Telethon Institute of Genetics and Medicine
David Fajgenbaum, MD
University of Pennsylvania
Anne Heatherington, PhD
Takeda Pharmaceuticals
Sponsors
The Winner’s Circle
Speakers
Jeong Ho Lee, MD, PhD
Korea Advanced Institute of Science and Technology
Adrian Krainer, PhD
Cold Spring Harbor Laboratory
Not Born This Way
Jeong Ho Lee, Early-Career Scientist winner of the 2020 Innovators in Science Award, discussed his work studying how somatic cell mutations—mutations that occur after development, during the normal process of cell division—result in rare neurological diseases caused by somatic cell mutations in the brain. Much of the recent boom in work on genetic diseases has focused on germline mutations. Because these mutations occur early in embryonic development, they show up in many types of cells throughout the body and are passed on to future offspring. These rare germline mutations can often be identified by sequencing the genomes of cells in easily accessible tissues, such as blood or skin. With advances in next-generation sequencing, “it’s become much easier to identify the germline mutations coding for many rare neurological disorders,” said Lee.
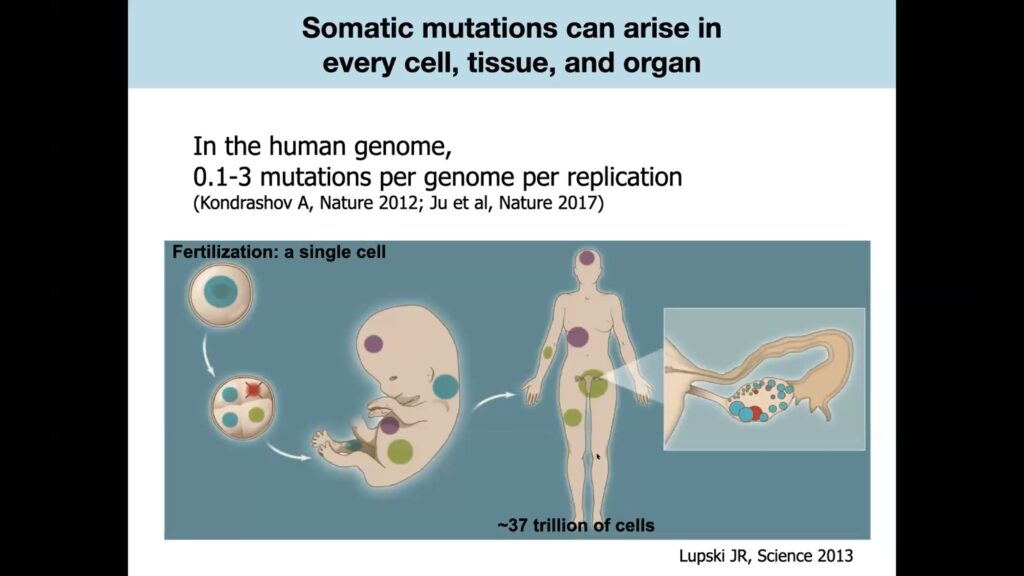
Nonetheless, germline mutations account for only a minority of rare neurological disorders. For example, 98% of epilepsy cases cannot be explained by germline mutations.
“We hypothesized that somatic cell mutations may be responsible for these unexplained neurological [diseases],” said Lee.
Somatic cell mutations occur during the ordinary cell division process that takes place billions of times in developing embryos, and continues to occur throughout life as somatic, or non-gamete, cells turn over.
DNA replication isn’t perfect, and human cells average 0.1 to 3 mutations per genome every time they divide. Lee theorized that a patient who hadn’t inherited an epilepsy-causing germline mutation might instead acquire somatic cell mutations in a subset of brain cells during development or later in life. If that happened, the mutation would only show up in the affected area of the brain, not in any other cells of the body.
One treatment for certain types of epilepsy is to resect the portion of the brain causing the seizures. Lee and his colleagues took samples of the brain tissue resected in these operations, along with blood samples from the same patients, and performed deep DNA sequencing to identify somatic mutations that occurred only in the affected brain tissue, not in the blood. They identified such mutations, including ones unique to genes involved in motor nerve activity, in 30% of the patients. When the scientists introduced the same mutations into a small percentage of neurons in developing mice, the animals developed epilepsy.
Next, the investigators looked at brain tumors, which can also cause epilepsy. One rare brain tumor type involves both glial cells and neurons, triggering epilepsy. Sequencing genetic information from cells in the tumors revealed that in 46% of affected patients, the glial cells and the neurons in the tumor shared an identical mutation.
“It means that the…neural stem cell already contained this… mutation,” said Lee, “and differentiated into the neuron and the glial cell.”
That could explain the high rate of disease recurrence in patients with these tumors; even if surgeons remove the entire tumor, the mutant stem cells might continue to produce more defective neurons and glial cells, which could then seed the growth of a new tumor.
To confirm that, Lee’s team collected an additional round of samples, this time sequencing cells not only from resected brain tumors and blood, but also from the subventricular zone in each patient’s brain, an area rich in undifferentiated neural stem cells. They found the tumor-associated mutation in the subventricular zone samples as well as the tumors, indicating that the error occurred in the neural stem cells, whose neuronal and glial descendants then migrated to where the tumor grew.
The researchers are also looking at neurodegenerative disorders such as Alzheimer’s disease.
“We hypothesized that somehow brain somatic mutation maybe accumulates over aging, and maybe associates with [Alzheimer’s disease development],” said Lee.
By performing deep sequencing on brain tissues from patients with and without Alzheimer’s disease, he and his colleagues identified somatic mutations unique to the diseased brains, supporting their theory.
In addition to identifying the underlying mechanisms behind neurological diseases, Lee is trying to help patients in other ways. In one effort, he has begun providing his results to clinicians to use in genetic counseling. Because conditions caused by somatic mutations aren’t heritable, while those caused by germline mutations are, patients who might be considering having children need to know which category they’re in.
The investigators are also trying to find ways to repair or mitigate the effects of somatic mutations in the brain, but it’s a tall order.
“Even if we found a molecular genetically validated target in the patient’s brain, it would be very difficult to develop a traditional drug to penetrate the blood-brain barrier and regulate the target,” Lee explained.
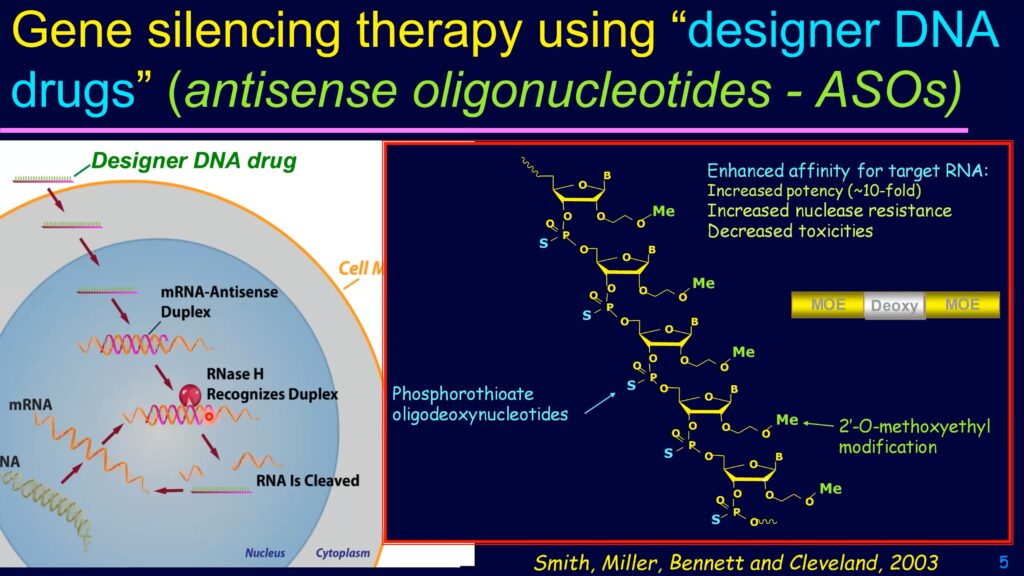
Instead, he’s hopeful that chemically modified strings of nucleic acids, called antisense oligonucleotides, will be able to target the somatic mutations he’s identified.
“I believe in the next five, ten, or twenty years, we probably can solve a lot of the rare neurological disorders,” he said.
Different Diseases, Different RNA Splices
Adrian Krainer won the 2020 Innovators in Science Senior Scientist Award, recognizing years of work spent developing treatments for rare diseases. Krainer and his colleagues were the first to develop an effective drug to treat spinal muscular atrophy. Affecting about 1 in 10,000 people worldwide, spinal muscular atrophy is an inherited genetic disease caused by a defect in the SMN1 gene. SMN1 encodes the SMN protein, which is essential for motor neuron survival. Patients with the mutation experience progressive loss of motor neurons, leading to loss of muscle control and, in most forms of the disease, early death.
Another gene, SMN2, also encodes the SMN protein, but cells usually splice out one of the protein coding sequences, or exons, from the SMN2 messenger RNA, preventing it from making the full-length protein. As a result, 80-90% of the protein translated from SMN2 RNA is truncated, nonfunctional, and rapidly degraded by the cell. Krainer reasoned that preventing the exon-skipping event might allow patients’ unmutated SMN2 genes to produce more functional SMN protein, overcoming the deficit caused by their mutated SMN1 genes. To do that, his team turned to antisense oligonucleotides, which encode the complementary, or antisense, sequence of a specific RNA target. When introduced into a cell, the antisense oligonucleotide binds specifically to its target sequence, triggering various cellular responses.
By designing an antisense oligonucleotide that altered the splicing of SMN2 messenger RNA, the researchers were able to get SMN1-mutant cells to produce more functional SMN protein. Subsequent preclinical and clinical trials proved that their antisense oligonucleotide also works in spinal muscular atrophy mouse models and in patients, respectively, significantly mitigating their motor neuron losses.
“Therefore this is a way that allows them to make closer to normal levels of a functional SMN protein in the presence of this drug,” said Krainer.
The oligonucleotide, now sold as nusinersen (Spinraza), was approved in the US in 2016 and the EU in 2017. Over 11,000 patients now receive it worldwide.

Based on the success of nusinersen, Krainer and his colleagues have begun looking at other RNA processing events to target with antisense oligonucleotides. One project focuses on familial dysautonomia, an inherited genetic disorder that affects only 310 known patients worldwide. These individuals have profound defects in their sensory neurons and autonomic nervous system, leading to symptoms that range from insensitivity to pain to difficulty swallowing.
“It is a very severe disease, a rare disease with a complex set of symptoms,” said Krainer, adding that “median survival is about 40 years of age.”
The condition is caused by a mutation in the gene for a protein called ELP1. As in SMN2, the mutation causes one exon of the gene’s messenger RNA to be spliced out, leading to a loss of functional ELP1 protein.
“So, we started targeting this aberrant splicing event using the same screening strategy and the same chemistry that we used…for spinal muscular atrophy,” said Krainer.
That effort identified an antisense oligonucleotide that can reverse the ELP1 RNA splicing defect in cultured cells from patients, as well as a transgenic mouse model.
“We feel that this is ready for clinical development; it is a challenge, though, because of the rarity of this disease,” said Krainer.
With only a few hundred patients in the US and Israel, the market for familial dysautonomia therapies is minuscule, and effective screening of potential carriers of the affected gene has led to very few new patients being born.
Not all RNA splicing-related diseases are rare, though. Work by several researchers has shown that in at least some cases, a change in the splicing of messenger RNA can help cancer cells grow. Alternatively, spliced forms of the messenger RNA for the PKM gene can produce two different isoforms of the metabolic enzyme pyruvate kinase. PKM1 predominates in normal adult tissues, while tumors and some developing tissues favor PKM2 production.
Using the same approach that worked in their rare disease work, Krainer’s team screened antisense oligonucleotides and identified candidates that bound the PKM messenger RNA and directed its splicing to favor PKM1 protein production. Putting these oligonucleotides into hepatocellular carcinoma cells causes the cells to shift their metabolism and slow their growth. In a mouse model of hepatocellular carcinoma, injecting the antisense oligonucleotides led to a significant reduction in tumor growth compared to control animals treated with saline solution.
Further Reading
Lee
Lee JH, Lee JE, Kahng JY, et al.
Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations
Nature. 2018 Aug;560(7717):243-247.
Lee, JH, Lee JH.
The Origin-of-Cell Harboring Cancer-Driving Mutations in Human Glioblastoma
Nat Med. 2015 Mar 23; 21(4):395-400.
Yoon, Seon-Jin, Junseong Park, Dong-Su Jang, Hyun Jung Kim, Joo Ho Lee, Euna Jo, Ran Joo Choi, et al.
Glioblastoma Cellular Origin and the Firework Pattern of Cancer Genesis from the Subventricular Zone
Nat Commun. 2019 July 12; 10(1):3090.
Krainer
Hua Y, Sahashi K, Rigo F, et al.
Peripheral SMN Restoration Is Essential for Long-Term Rescue of a Severe SMA Mouse Model
Nature. 2011 Oct 6; 478(7367):123-126.
Sinha R, Kim YJ, Nomakuchi T, et al.
Nucleic Acids Res. 2018 Jun 1; 46(10):4833-4844.
Wang Z, Jeon HY, Rigo F, Bennett CF, Krainer AR.
Manipulation of PK-M Mutually Exclusive Alternative Splicing by Antisense Oligonucleotides
Open Biol. 2012 Oct; 2(10):120133.
Bennett CF, Krainer AR, Cleveland DW.
Antisense Oligonucleotide Therapies for Neurodegenerative Diseases
Annu Rev Neurosci. 2019 Jul 8; 42: 385-406.
Muscle and Brains
Speakers
Annemieke Aartsma-Rus, PhD
Leiden University Medical Center
Don Cleveland, MD, PhD
University of California, San Diego
The Kindest Cut
Annemieke Aartsma-Rus began the meeting’s third session with a presentation about her group’s efforts to address Duchenne muscular dystrophy with antisense oligonucleotides. While Krainer’s approach to rare diseases focuses on conditions where an exon needs to be added back into a messenger RNA, Aartsma-Rus described a case where it’s better to remove one.
Duchenne muscular dystrophy is an X-linked genetic disorder. In most cases, a mutation in the dystrophin gene shifts the messenger RNA’s reading frame, causing translation of the dystrophin protein to fail.
“Patients become wheelchair dependent around the age of 12, need assisted ventilation around the age of 20, and generally die in the second to fourth decade of life,” said Aartsma-Rus.
A related but milder disorder, Becker muscular dystrophy, also involves a deletion in the dystrophin gene but doesn’t shift the messenger RNA’s reading frame. As a result, patients with Becker muscular dystrophy produce partially functional dystrophin and exhibit a slower disease progression.

Looking at the affected DNA and RNA sequences, Aartsma-Rus reasoned that most Duchenne muscular dystrophy patients could make Becker-like dystrophin, if their cells could simply skip the affected exon in their dystrophin messenger RNA. To test that, she and her colleagues developed chemically modified antisense oligonucleotides that would remain stable in blood and tissues, and began testing them as potential drugs. By designing an oligonucleotide that targeted RNA splicing, the team restored dystrophin expression in cultured cells carrying a Duchenne muscular dystrophy mutation.
The researchers discovered a potential roadblock in a mouse model: antisense oligonucleotides injected into the animals’ tail veins were absorbed almost entirely by the liver and kidneys. The investigators could inject the molecules directly into muscles instead, but that clearly wouldn’t be a practical way to deliver treatment to patients.
“We have over 700 different muscles, and you’ll have to treat patients repeatedly,” said Aartsma-Rus, “so local injection of each and every muscle weekly or even monthly is likely not realistic.”
However, in a mouse model of Duchenne muscular dystrophy, the team discovered that oligonucleotides injected into the animals’ tail veins were absorbed into muscles ten times better than they had been in wild-type mice.
“The first time we thought we’d made a mistake, so we repeated it a couple of times,” said Aartsma-Rus, “but every time we saw that there was higher uptake by the dystrophic muscle than the healthy muscle.”
Dystrophin deficiency causes muscle cells to become more permeable, leading to leakage of cellular components, but this leakage works in both directions; the dystrophic cells readily absorbed oligonucleotides that healthy cells excluded.
Flush with this preclinical victory, the team began setting up clinical trials in 2007. The initial multi-center, open-label trial found that the antisense oligonucleotides caused no serious side effects, and eight of the twelve patients tested saw their conditions remain stable throughout the trial. To evaluate efficacy, the investigators moved into a phase 2b trial, which continued to show dose-dependent effectiveness in treated patients. However, a larger phase 3 trial yielded disappointment, with no significant difference in outcomes between treated and control patients.
“So, what happened [to explain why] we see these beneficial effects in the phase 2 trial, but in the phase 3 trial we see no effect?” Aartsma-Rus asked.
Analyzing the results and the disease further, she realized that the trials were built on the flawed assumption that the patients’ progression would be linear. Instead, they realized that younger patients tend to remain stable for an extended period, followed by a rapid decline later in life. By mixing different ages in the phase 3 trial, patients with worse disease symptoms likely masked any treatment benefits in those with milder symptoms.
Looking at the trial’s failure, Aartsma-Rus concluded that she and her colleagues should have opened discussions with regulatory agencies sooner, and studied the natural history of the disease more thoroughly, before initiating the phase 3 study. Unfortunately, the expensive late-stage failure has soured companies on further clinical development of exon-skipping antisense oligonucleotides for Duchenne muscular dystrophy. Aartsma-Rus has since focused on preventing such an outcome in the future.
“Now we have an open dialogue with academics, with patients, with regulators in the EU, and it is also starting in the US, developing new outcome measures,” she said, adding that “future trials will be better.”
Batting for Lou Gehrig
“These DNA drugs were ten to fifteen times the size of a typical drug, and they’re heavily charged,” said Cleveland, “so the pharmacology textbooks all said that there was no uptake mechanism that would permit them to be efficiently taken up [by neurons].” Nonetheless, he continued, “we tried it anyway, and it turns out that the cells of the nervous system hadn’t read the textbooks.”
Injecting the antisense oligonucleotides into the cerebrospinal fluid of mice genetically modified to develop severe ALS doubled the animals’ survival times.
While the superoxide dismutase defect was the first ALS mutation discovered, the most common cause of the inherited form of the disease is a mutation in a gene called ORF72, which inserts extra nucleotides into a non-coding region of the gene. This causes defective messenger RNA to accumulate, killing neurons because of a lack of functional ORF72 gene products and the accumulation of toxic byproducts of the altered gene. Antisense oligonucleotides targeting the defective RNA, however, inhibit its accumulation without reducing the production of working ORF72 gene products in cultured cells.
In an animal model of the ORF72 defect, the results were even more impressive.
“We dosed these animals [with the antisense oligonucleotides] at the age of disease onset and asked what happens, and the answer is we prevented further disease development for the life of those animals with a single dose injection applied at the initial signs of disease,” said Cleveland.
His team initiated clinical trials on this therapy in 2018, just seven years from the date when researchers had first published the data showing the ORF72 mutation caused ALS.
Although it’s an important target for research, inherited ALS accounts for only 10% of the disease’s total cases. In 90% of patients, the condition develops spontaneously due to somatic cell mutations later in life. Many of these cases involve mutations in the TDP-43 gene, which encodes a nuclear protein that regulates Stathmin-2, which in turn plays a critical role in regulating the cytoskeleton in neurons. TDP-43 normally binds the Stathmin-2 precursor RNA and ensures that it gets spliced properly into messenger RNA. Mutations that inactivate TDP-43 cause a loss of functional Stathmin-2, which is a hallmark of sporadic ALS.
Using cultured neurons, Cleveland and his colleagues found that a properly designed antisense oligonucleotide could compensate for the loss of TDP-43 activity, restoring normal RNA splicing and Stathmin-2 expression.
“This now enables a strategy for therapy for sporadic ALS,” said Cleveland.
If the result holds in other preclinical models, he expects to take that approach into clinical trials in 2023.
Besides correcting specific defects within a cell, antisense nucleotides can potentially redirect a cell’s fate entirely. That’s the central theme of another project in Cleveland’s lab, in which the team is causing astrocytes to change their identities. Astrocytes are companion cells in the nervous system that arise from the same stem cells as neurons. Using antisense oligonucleotides, the investigators can suppress two genes that direct cells into the astrocyte lineage, causing them to become neurons instead. Cleveland is initially focusing on treating Parkinson’s disease with this approach, but he explained that “this…conversion of astrocytes into replacement neurons may be broadly applicable for neurogenic disease.”
Further Readings
Aartsma-Rus
Aartsma-Rus A, Ginjaar IB, Bushby K.
The Importance of Genetic Diagnosis for Duchenne Muscular Dystrophy
J Med Genet. 2016 Mar;53(3):145-151.
Verhaart IEC, Aartsma-Rus A.
Therapeutic Developments for Duchenne Muscular Dystrophy
Nat Rev Neurol. 2017 Jul;15(7): 373-386.
Verhaart IEC, Robertson A, Wilson IJ, et al.
Orphanet J of Rare Dis. 2017 Jul 4;12(1):124.
Cleveland
Cleveland DW, Bruijn LI, Wong PC et al.
Mechanisms of Selective Motor Neuron Death in Transgenic Mouse Models of Motor Neuron Disease
Neurology. 1996 Oct;47(4 suppl 2): S54-61; discussion S61-62.
Saberi S, Stauffer JE, Jiang J, et al.
Acta Neuropathol. 2018 Mar;135(3):459-474.
Vechio JD, Bruijn LI, Xu Z, et al.
Ann Neurol. 1996 Oct; 40(4):603-10.
A Spectrum of Phenotypes
Speaker
Huda Zoghbi
Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital
Maybe Not So Rare
Huda Zoghbi gave the meeting’s keynote presentation, which covered her work on Rett syndrome. Caused by spontaneous mutations in the MECP2 gene on the X chromosome, Rett syndrome is a progressive neurodegenerative disease that primarily manifests itself in girls. MECP2 is critical for gene regulation in neurons. Because females carry two copies of the X chromosome and an inactivate one in each cell, an inactivating mutation in MECP2 impairs the function of 50% of the affected individual’s neurons. That manifests itself as a rapid regression in motor and cognitive abilities by age two.
In boys, who have only one X chromosome, inactivation of MECP2 is generally lethal before age two. They don’t live long enough to develop the classic symptoms of Rett syndrome. However, recent work has revealed that some males acquire mutations that cause less severe defects in MECP2.
“What we’ve learned is when people carry milder mutations, we will see milder phenotypes, such as mild learning disability with…neuropsychiatric features,” said Zoghbi.
These individuals’ phenotypes can range from autism to hyperactivity or schizophrenia, but most die by middle age due to neurodegeneration. Females with mild defects in MECP2 show non-random inactivation of their X chromosomes, favoring the healthy copy of the gene and enabling them to develop and live normally. Some patients also have duplications in their MECP2 genes, often leading to severe neurological problems and premature death.
To understand the mechanisms driving Rett syndrome, Zoghbi and her colleagues developed a series of genetically modified mice carrying various duplications or mutations in MECP2. Consistent with the findings in humans, these animals display a spectrum of phenotypes depending on the severity of their MECP2 disruptions.
“The brain is very sensitive to the activities and functions of this protein, and we’ve done a lot of studies on both the loss and the gain models,” said Zoghbi.
She and her colleagues found that all types of neurons require functional MECP2 to operate normally.

Next, Zoghbi and her colleagues tried inactivating MECP2 in excitatory and inhibitory neurons separately. They found that in both cases, animals developed obesity, lost motor coordination, and died young. However, targeting MECP2 only in inhibitory neurons led to more learning and social defects in the animals, while inactivating it only in excitatory neurons caused more anxiety and tremors. These phenotypes represent the downstream effects of the genes MECP2 would normally regulate.
“Given that it’s important for practically every cell, really there’s two major ways you can think of treating this disorder, either gene replacement therapy…or perhaps exploring modulation of the [MECP2 regulatory] circuit,” said Zoghbi.
Taking the latter approach, the investigators implanted electrodes into the brains of mice to deliver small electrical pulses. This type of deep brain stimulation, which has been shown to reverse many types of neuronal signaling and development defects, is already approved for human treatment of several neurological disorders. Stimulating the brains of Rett syndrome model mice leads to significant recovery in their learning, memory, and motor abilities that persists for weeks after treatment.
“It was really quite a dramatic rescue in that all these phenotypes normalize, and their normalization…lasted for several weeks,” said Zoghbi.
The treated animals’ neurons also displayed gene expression patterns similar to wild-type animals, whereas untreated animals showed significant gene dysregulation.
“The Rett brain, at least in mice, is responsive to neuromodulation,” said Zoghbi.
Looking at the MECP2 gene itself, Zoghbi’s team identified regulatory sequences that control its expression level. Altering these sequences to increase or decrease the amount of MECP2 expressed in mice underscored their earlier findings, showing that even modest changes in MECP2 levels led to detectable neurological phenotypes.
Like other speakers at the meeting, Zoghbi and her colleagues are also exploring the potential of antisense oligonucleotides as therapies. That approach seems especially promising for patients with duplications in MECP2 that lead to overexpression of the gene. In mice that recapitulate this condition, the researchers found that treatment with antisense oligonucleotides against MECP2 could reduce the amount of functional protein in neurons down to wild-type levels. The treatment reversed the animals’ motor defects.
Titrating the antisense oligonucleotide dosage also revealed that even modest decreases in excess MECP2 can lead to major improvements in symptoms.
“If you can even partially decrease the protein…you will probably rescue quite a bit of the features of the disease,” Zoghbi said. She added, “I’ve really never worked with a protein that is so exquisitely sensitive to the levels.”
Further Readings
Zoghbi
Fagiolin M, Patrizi A, LeBlanc J, et al.
Neuroscience. 2020 Oct 1;445:190-206.
Neul JL, Fang P, Barrish J, et al.
Specific Mutations in Methyl-CpG-Binding Protein 2 Confer Different Severity in Rett Syndrome
Neurology. 2008 Apr 15;70(16):1313-1321.
.
The Way Forward
Panelists
Graciana Diez-Roux, PhD
Telethon Institute of Genetics and Medicine
David Fajgenbaum, MD
University of Pennsylvania
Anne Heatherington, PhD
Takeda Pharmaceuticals
Silver Linings
The meeting’s general session concluded with a panel discussion led by Brad Margus, co-founder and CEO of Cerevance. With a background in business, Margus moved into rare disease drug development after his daughter was diagnosed with ataxia-telangiectasia, a genetic disorder that causes neurodegeneration and immune dysfunction. The panel also featured Graciana Diez-Roux, chief scientific officer at the rare disease-focused Telethon Institute; David Fajgenbaum, a physician-scientist who both studies and suffers from Castleman syndrome; and Anne Heatherington, a data scientist for Takeda Pharmaceuticals with extensive experience studying Duchenne muscular dystrophy.
Panel members discussed the need for improvement in collaborations between patients and researchers.
“There is a lot of miscommunication within the rare disease research space, [but] I think there’s been a really great trend for groups like Takeda and others toward engaging patients in the research process,” said Fajgenbaum, adding that “I also think clinicians can really be a part of this.”
Besides improving clinical trial recruitment, involving patients more directly in research can have far-reaching benefits for scientists.
“It’s incredible how our PhD students, when they have the chance [to interact] with the patients and [get] to know the patients’ organizations…how their motivation and their love for what they do changes,” Diez-Roux said.
Besides increasing collaborations between patients and scientists, all of the panelists endorsed the need for strong, well-defined partnerships with pharmaceutical companies. Margus described his company’s efforts to improve data collection and sharing for ataxia-telangiectasia, which included building a system that uses wearable devices to collect movement data from patients around the clock.
“The data [are] truly owned by the families and the community, and we can make decisions about sharing the data with academics or any researcher in the world in a matter of days,” said Margus.
Good partnerships require more than just good databases, though. Academic researchers accustomed to independent, curiosity-driven experimental design and flexible deadlines sometimes have trouble accommodating pharmaceutical companies’ urgent, goal-directed needs.
“I think the model has to be somewhere between…the industry knowing how to deal with the academic research and academic researchers being open to notice that industries have…different goals in some respects,” said Diez-Roux.
The group also discussed the impact of the COVID-19 pandemic. In the short term, of course, the global shutdown caused by the SARS-CoV-2 virus has halted or delayed many rare disease studies. However, panelists agreed that some of the innovative approaches developed for the pandemic response could transform many aspects of rare disease research in the future.
“We have been very involved in a lot of the COVID alliances, and have been steeped in novel ways of working,” said Takeda’s Heatherington.
As an example, she pointed to the company’s involvement in multi-corporation consortia to develop new therapies and even entirely new platforms for therapies.
“That’s a real breakthrough in terms of how we do our business, that extent of collaboration for [competitors to] come together,” Heatherington continued.
At the same time, “the public is realizing more how important research is, and this goes for COVID, but I think it goes for all diseases,” said Diez-Roux. Both she and Heatherington also pointed out that the pandemic has underscored the potential tradeoffs between speed and safety in therapeutic development, and highlighted the importance of oversight in clinical trials.
The meeting concluded with a virtual poster session, featuring rapid-fire presentations of some of the newest research in rare diseases and offering attendees the ability to interact with the presenters directly. Like the other presentations, the posters represented the diversity and enthusiasm of rare disease researchers.
“What makes me optimistic is the passion and the knowledge…and the fact that we have people that are so dedicated to rare diseases,” said Heatherington.
Further Readings
Heatherington
Bhattacharya I, Manukyan Z, Chan P, et al.
Clin Pharmacol Ther. 2016 Oct;100(4):330-332.
Dwivedi G, Fitz L, Hegen M, et al.
CPT: Pharmacometrics Syst Pharmacol. 2014 Jan 8;3:e89.
Tiwari A, Kasaian M, Heatherington AC, et al.
MAbs. 2016 Jul;8(5):983-990.
Fajgenbaum
Leurs A, Gnemmi V, Lionet A, et al.
Front Immunol. 2019 Jun 28;10:1489.
Oksenhendler E, Boutboul D, Fajgenbaum D, et al.
The Full Spectrum of Castleman Disease: 273 Patients Studied over 20 Years
Br J Haematol. 2018 Jan;180(2):206-216
Soudet S, Fajgenbaum D, Delattre C, et al.
Curr Res Transl Med. 2018 Sep;66(3):83-86.
Diez-Roux
Diez-Roux G, Ballabio A.
Annu Rev Genomics Human Genet. 2005;6:355-379.
Diez-Roux G, Banfi S, Sultan M, et al.
A High-Resolution Anatomical Atlas of the Transcriptome in the Mouse Embryo
PLoS Biol. 2011 Jan 18;9(1):e1000582.
Gatto F, Rossi B, Tarallo A, et al.
Sci Rep. 2017 Nov 8;7(1):15089.