Equivalence of Complex Drug Products: Scientific and Regulatory Challenges
Reported by:
Jennifer Cable
Presented by:
The Non Biological Complex Drugs Working Group (NBCD WG)
The New York Academy of Sciences
Overview
Biotechnology and nanotechnology have given rise to a growing number of innovator-driven complex drug products and their copy versions. Biologics exemplify one category of complex drugs, but there also exist non-biological complex drug products (NBCDs), including many nanomedicines such as iron-carbohydrate complexes, several drug carrying liposomes or emulsions, and glatiramoids. These products are difficult to characterize—often the key features for efficacy and safety are not well understood. These factors pose unique challenges for pharmaceutical companies and regulatory agencies when comparing generic complex drugs to their branded counterparts.
On November 9, 2016, representatives from academia, industry, and regulatory agencies convened at the New York Academy of Sciences for the symposium Equivalence of Complex Drug Products: Scientific and Regulatory Challenges, an opportunity to discuss how to approach complex generics. Regulatory officials described how agencies approach evaluating complex generics for marketing approval, while industry speakers presented several examples that demonstrate the difficulties in characterizing complex drugs.
The symposium was presented by the Non Biological Complex Drugs Working Group, the Nanotechnology Characterization Laboratory, and the New York Academy of Sciences.
Speakers
Keynote Speaker
Lawrence Mayer, PhD
Jazz Pharmaceuticals
website | publications
Lawrence Mayer, senior vice president of discovery at Jazz Pharmaceuticals, has played a lead role in the discovery and development of a number of oncology drugs, several of which eventually achieved market approval. He has also held senior management positions at The Canadian Liposome Company and QLT, Inc. before joining the British Columbia (BC) Cancer Agency, where he established and directed the Health Canada-accredited Investigational Drug Program. Celator Pharmaceuticals, now a subsidiary of Jazz Pharmaceuticals, was formed in 2000 as a spin-out of Mayer’s laboratory at the BC Cancer Agency. Mayer has authored more than 250 scientific publications and has more than 35 patent families awarded or pending. Mayer received his BS in both chemistry and biology, summa cum laude, from Wartburg College and his PhD in biochemistry from the University of Minnesota.
Speakers
Daan J.A. Crommelin, PhD
Utrecht University
website | publications
Daan Crommelin is professor emeritus at the Department of Pharmaceutics at Utrecht University. Until December 2011 he was scientific director of the Dutch Top Institute Pharma in Leiden. He is adjunct professor at the Department of Pharmaceutics and Pharmaceutical Chemistry at the University of Utah. Crommelin is co-founder of OctoPlus, a Leiden based company specialized in the development of pharmaceutical (mainly protein based) product formulations and advanced drug delivery systems. He published extensively and is on the editorial board of 10 peer reviewed journals in the pharmaceutical sciences. He is Editor-in-Chief of the AAPS book series Advances in the Pharmaceutical Sciences. He advises venture capital groups and acts as consultant. He chairs the UCAB Foundation: the Utrecht Center of Excellence for Affordable Biotherapeutics, a WHO supported initiative. He chaired the Board of Pharmaceutical Sciences of the International Pharmaceutical Federation (F.I.P.), was chair of the organizing committee of the Pharmaceutical Sciences World Conference 2007 in Amsterdam. He is past president of the European Federation of Pharmaceutical Sciences (EUFEPS) and past vice-chair of the scientific advisory board of the European Innovative Medicines Initiative (IMI).
Beat Flühmann, PhD
Vifor Pharma Ltd
website | publications
A pharmacist by training, Beat Flühmann holds a PhD in molecular biology. In his current position Flühmann serves as global lead on non-biological complex drugs at Vifor Pharma Ltd Switzerland, with a main interest in regulatory aspects of nanomedicines. Previously, Flühmann led a global multidisciplinary research and development team at Roche/DSM nutritional products developing novel compounds for the prevention and treatment of diabetes. He also is a steering committee member of the Non-Biological Complex Drugs Working Group hosted at the nonprofit Lygature, a group set up to discuss science-based approval and post-approval standards to ensure patient safety and benefit with non-biological complex drugs. The working group engages in activities to publish and discus scientific evidence with authorities, experts, health care providers, and is involved in scientific education and training to relevant stakeholders. Flühmann also sits on the advisory board of the GoNanoBioMat project, a multinational research project in the field of nanomedicine.
Joseph Glajch, PhD
Momenta Pharmaceuticals, Inc.
website | publications
Joseph L. Glajch is director of analytical development at Momenta Pharmaceuticals. He received his AB in chemistry at Cornell University and PhD in analytical chemistry at the University of Georgia under L.B. (Buck) Rogers. He has held technical and R&D management positions at DuPont, Bristol-Myers Squibb, Certus, and Momenta, with an emphasis on HPLC column and method development and pharmaceutical development and analysis. He has served as president and program chairman of the Analytical Division of the American Chemical Society and the Chromatography Forum of the Delaware Valley, as well as program chair of the Gordon Conference on Analytical Chemistry. He has served on the editorial advisory boards of Analytical Chemistry, the Journal of Chromatography, and LC/GC. He is a member of the USP Expert Committee on General Chemical Analysis and has over 40 publications, is co-author of three books, and holds six patents on HPLC column materials and medical imaging agents.
Elwyn Griffiths, PhD, DSc
World Health Organization and Health Canada
publications
Elwyn Griffiths is currently a consultant in vaccines and biotherapeutics to WHO and a member of the WHO Expert Committee on Biological Standardization. He is also a member of the Clinical Trials, Biologicals and Vaccines Expert Advisory Group of the UK Commission on Human Medicines and of the Expert Advisory Group on Biological and Biotechnology products of the British Pharmacopoeia Commission.
In 2011, Griffiths retired as director general, biologics and genetic therapies directorate of Health Canada, an organization he joined in 2003 upon retirement from the World Health Organization. Griffthis holds a PhD and a DSc degree from the University of Wales. In 1980, he became a senior member of staff at the UK National Institute for Biological Standards and Control, and in 1994 was appointed chief of biologicals at WHO. From 1994 until the end of 2002, Griffiths was responsible for WHO’s program for providing international written and physical standards for vaccines, blood products and biotherapeutics.
Iris Grossman, PhD
Teva Pharmaceuticals
website | publications
Iris Grossman, VP, global head of the Personalized & Predictive Medicine (PPM) and Big Data Analytics unit for Teva Global R&D, is currently charged with defining and implementing Teva’s global PMP strategy, covering both discovery and development R&D programs. Israel’s leading financial magazine, Globes Magazine, selected Grossman as one of the country’s top 40 professionals under 40 years of age in 2013.
Prior to joining Teva, Grossman was CEO and president of the pharmacogenetics management consultancy IsraGene Ltd.. This followed several years of spearheading pipeline pharmacogenetic programs for industry and academia as director of pharmacogenetics at Cabernet Pharmaceuticals Inc. Grossman moved into consultancy having been responsible for running large-scale pharmacogenetic programs at GlaxoSmithKline, with an emphasis on infectious and neurological diseases.
Grossman received her PhD from the Technion–Israel Institute of Technology, where her research project, conducted in collaboration with the Weizmann Institute for Science, investigated pharmacogenetic markers of multiple sclerosis treatment response.
Wenlei Jiang, PhD
Office of Research and Standards, Office of Generic Drugs, US Food and Drug Administration
website | publications
Wenlei Jiang is currently a senior science advisor in the Office of Research and Standards (ORS)/Office of Generic Drugs (OGD)/Center for Drug Evaluation and Research (CDER). She is mainly responsible for coordinating post-market generic drug safety investigation, representing ORS on OGD’s new international harmonization activities, and developing opportunities for scientific outreach. Previously, she served as the acting deputy director of ORS, where she provided oversight on Generic Drug User Fee Act (GDUFA) regulatory science research programs. She used to work in the Division of Chemistry, OGD to review the chemistry and manufacturing control (CMC) sections of ANDAs. Prior to joining FDA, she was at Novartis Pharmaceutical Corporation where her responsibilities included formulation development of conventional liquid and solid dosage forms, as well as advanced parenteral drug delivery systems. She received her PhD in pharmaceutics and pharmaceutical chemistry from Ohio State University in 2001.
Scott McNeil, PhD
Nanotechnology Characterization Laboratory
website | publications
Scott McNeil serves as director of the Nanotechnology Characterization Laboratory (NCL) for Leidos Biomedical Research and Frederick National Laboratory for Cancer Research, where he coordinates preclinical characterization of nanotech cancer therapeutics and diagnostics. At the NCL, McNeil leads a team of scientists responsible for testing candidate nanotech drugs and diagnostics, evaluating safety and efficacy, and assisting with product development. McNeil is a member of several working groups on nanomedicine, environmental health and safety, and other nanotechnology issues. He is an invited speaker to numerous nanotechnology-related conferences and has several patents pending related to nanotechnology and biotechnology. He is also a vice president at Leidos Biomedical Research.
McNeil’s professional career includes tenure as an Army officer, with tours as chief of biochemistry at Tripler Army Medical Center, and as a combat arms officer during the Gulf War. He received his bachelor’s degree in chemistry from Portland State University and his doctorate in cell biology from Oregon Health Sciences University.
Kouros Motamed, PhD
NantBioScience, Inc.
website | publications
Kouros Motamed, PhD has been the Director of Drug Development at NantBioScience, Inc. since April 2016. Prior to that, he has served as VP of Strategic Alliances and Clinical Communications and VP of Clinical Development and Nanomedicine at Sorrento Therapeutics from 2013 to 2016. He has also served as a co-founder and CSO/CTO of Igdrasol, Inc. and Biomiga Diagnostics start-up companies from 2011–2013. Dr. Motamed has also served as the MOA and Molecular Biology Group Head at Celgene Corp. and Abraxis BioScience Inc. from 2007–2011. Dr. Motamed held an Assistant Professorship position in the Department of Pathology and Vascular Biology Center at Georgia Health Sciences University from 2002–2007. He has over 30 original publications in peer-reviewed journals, over 50 conference presentations and has 5 issued patents. He has served on the Editorial Board of Journal of Nanomaterials & Molecular Nanotechnology since 2013. Dr. Motamed received a BS degree in Biology from University of San Francisco and a PhD degree from the University of California, Davis in Microbiology.
Stefan Mühlebach, PhD
Vifor Pharma Ltd
website | publications
Stefan Mühlebach, regulatory science lead for non-biological complex drugs at Vifor Pharma–Fresenius Medical Care Renal Pharma Ltd. (Switzerland), chairs the Non-Biological Complex Drugs (NBCDs) Working Group at Lygature, a nonprofit, private-public partnership in the Netherlands. He is a university professor, medical faculty member, and member of the clinical pharmacy & epidemiology unit at the Department of Pharmaceutical Sciences at the Natural Sciences Faculty (Switzerland). Research activities, graduate and postgraduate teaching cover topics in pharmacology, clinical nutrition, hospital pharmacy, and regulatory sciences. He has authored over 100 peer-reviewed papers and several book chapters. From 1980 to 2005, he served as a chief hospital pharmacist in Switzerland. He is an honorary member of the Swiss Association of Public Health Administration and Hospital Pharmacists and a board member of the Swiss Academy of Pharmaceutical Sciences. In 2008, he joined Vifor Pharma Ltd as chief scientific officer.
Chetan Pujara, PhD
Allergan
website | publications
Chetan Pujara is vice president, small molecule product development at Allergan, Plc. His organization is responsible for designing and developing pharmaceutical dosage forms intended for clinical trials and commercialization. SMPD develops topical sterile ophthalmic solutions, suspensions and emulsions, sustained-release ocular biodegradable implants, oral tablets/capsules, topical dermal gels & creams and other locally acting dosage forms. Prior to joining Allergan, Chetan was employed by Abbott Laboratories/Abbvie, where he held various positions in global pharmaceutical R&D, gaining experience in development of solid oral dosage forms and pediatric oral suspensions.
Pujara is also a member of the USP Dosage Forms Expert Committee and serves on the USP <771> Ophthalmic Preparation Expert Panel. He serves as a scientific advisor to the editors of Journal of Pharmaceutical Sciences and an adjunct professor in the Department of Industrial and Physical Pharmacy, Purdue University. Pujara has also contributed to the Physical Properties Working Group of Product Quality Research Institute.
Andre Raw, PhD
Center for Drug Evaluation and Research, US Food and Drug Administration
website | publications
Andre Raw received his BS from the Massachusetts Institute of Technology and his PhD in chemistry from the University of California at Berkeley. He 2001, he joined the FDA as a reviewer within the Office of Generic Drugs, where he is currently acting senior scientific and policy advisor in the Office of Lifecycle Drug Products in the Office of Pharmaceutical Quality. Raw was involved in the development of several FDA initiatives, including the Guidance on Pharmaceutical Solid Polymorphism in Abbreviated New Drug Applications (ANDAs), Regulations on Listing of Polymorph Patents in the “Orange Book,” and Question Based Review – Quality by Design (QbD) Initiative, QbD Example for Generic Modified Release Products, and Guidance for Industry: Pharmaceutical Solid Co-Crystals. He has also been active in addressing scientific and regulatory issues raised in citizen petitions. He was instrumental in FDA’s recent approval of generic versions of complex active ingredients including Lovenox (enoxaparin sodium), and Ferrlecit (sodium ferric gluconate complex in sucrose).
Stephan Stern, PhD, DABT
Nanotechnology Characterization Laboratory
website | publications
Stephan Stern is acting deputy director and senior principal scientist at the National Cancer Institute’s Nanotechnology Characterization Laboratory (NCL), located at the Frederick National Laboratory for Cancer Research in Frederick, Maryland. NCL assists in all phases of the nanomedicine drug development process, from early preclinical to late stage clinical trials, working with academic laboratories and the pharmaceutical industry. At NCL, Stern oversees nanomedicine pharmacology and toxicology. Stern’s research interests include novel drug formulation, bioanalytical method development, and pharmacokinetic modeling. Prior experience includes a postdoctoral fellowship at the University of North Carolina–Chapel Hill in the Division of Drug Delivery and Disposition, and curriculum in toxicology, and work within regulated areas of the pharmaceutical industry. He received his BS in biochemistry from the University of Rochester and his PhD in toxicology from the University of Connecticut at Storrs. Stern is a diplomate of the American Board of Toxicology.
Wyatt Vreeland, PhD
The National Institute of Standards and Technology
publications
Wyatt Vreeland earned his PhD in chemical engineering from Northwestern University, where he developed techniques for the electrophoretic separation of natural and synthetic polymers in microfluidic devices. Later, Vreeland joined National Institute of Standards and Technology as an National Research Council postdoctoral, where Vreeland focused on using liposomes to package chemicals and release those reagents at controlled times and locations in microfluidic systems. Additionally, during his postdoctoral fellowship he undertook the technical development of a field-portable electrophoresis system for forensic DNA analysis for the Department of Justice. Most recently, Vreeland’s research has focused on controlled synthesis and characterization of colloidal protein systems as can be encountered (usually undesirably) in many of today’s complex drug therapeutics. In 2015, Vreeland served as an U.S. State Department Embassy Science Fellow for the U.S. Embassy in Prague, Czech Republic, where he toured the country’s burgeoning biotechnology industry.
Gary West, MD
Azaya Therapeutics
website | publications
Gary West is currently a member of the board of directors of Azaya Therapeutics. A retired radiation oncologist, he graduated from the University of Colorado Medical School and did his radiation oncology residency at the University of New Mexico, where he was affiliated with New Mexico’s Los Alamos Meson Facility (LAMF), before completing his residency at the Joint Center for Radiation Oncology (JCRO), Harvard University. He completed a research fellowship at JCRO, where he investigated chemotherapy drug delivery in human tumor cells. While in the Air Force, he was chief of the radiation oncology department at Wilford Hall USAF Medical Center. He has also served as chief of staff at the University of Texas Health Science Center, Cancer Therapy, and Research Center (CTRC), in San Antonio and as a member of the CTRC board of governors. He is a retired member of the American Society of Clinical Oncologists and the American Society of Therapeutic Radiologists.
Elena Wolff-Holz, MD
Paul Ehrlich Institute
website | publications
Elena Wolff-Holz is medical assessor at the Paul-Ehrlich-Institut and a member of the Biosimilar Working Party (BMWP) of the Committee for Medicinal Products for Human Use (CHMP) and a national expert to the Oncology Working Party (OWP) of the CHMP, where she contributes to scientific advice procedures and the development of guidelines and reflection papers. Wolff-Holz spent 14 years in the biotech industry, where she held various positions in clinical development and medical marketing functions at Centocor Inc (now J&J) and Amgen in the U.S. and in Germany, before joining the German national regulatory agency. In the area of biotherapeutics, she is responsible for advising companies on drug development issues, reviewing applications for marketing authorization in the EU and assessing clinical trial applications. She is also a lecturer covering training of experts and executives from academia, regulatory bodies and biopharmaceutical industry. Wolff-Holz has an MD degree from Heidelberg University and a postdoctoral fellowship at Harvard Medical School.
Gillian Woollett, DPhil, MA
Avalere Health
website | publications
Gillian Woollett, SVP at Avalere, where she leads the FDA Policy and Regulatory Strategy Practice, an advisory services firm she launched in 2012 that supports clients throughout the healthcare system, from patients to biopharma companies and payers/providers. Woollett and her team translate into practical action all aspects of regulatory engagement strategy and policy development relevant to commercial success for multiple multi-national clients. Concurrently, she created the Avalere FDA Fellows Program to enable scientists to transition effectively into the policy environment with over 20 now having used this stepping stone to transition careers. Woollett earned her BA and MA in biochemistry from the University of Cambridge, and her DPhil in immunology from the University of Oxford. She was a postdoc in the Department of Molecular Biology at the University of Edinburgh, and at the Biomedical Research Institute, Rockville, MD funded by USAID.
Jon de Vlieger, PhD
Non Biological Complex Drugs Working Group and Lygature
website | publications
Jon de Vlieger obtained his doctoral degree in bio analytical chemistry from Vrije University (the Netherlands). In 2011, he joined Top Institute Pharma, an independent nonprofit organization based that catalyzes the development of new medical solutions for patients by driving public–private collaboration between academia, industry, and society. Recently, a merger between leading Dutch technology institutes Center for Translational Molecular Medicine and Top Institute Pharma formed Lygature. For Lygature, de Vlieger coordinates several international public private partnerships, including the Non Biological Complex Drugs Working Group, an international network of scientific and clinical experts from academia, industry and regulatory bodies, with expertise in many aspects of the development and evaluation of NBCDs. He is a co-editor of the book on NBCDs in the American Association of Pharmaceutical Scientists’ Advances in the Pharmaceutical Sciences Series.
Organizers
Melanie Brickman Borchard, PhD, MSc
The New York Academy of Sciences
Alison Carley, PhD
The New York Academy of Sciences
Daan J.A. Crommelin, PhD
Utrecht University
Wenlei Jiang, PhD
Office of Research and Standards, Office of Generic Drugs, US Food and Drug Administration
Scott McNeil, PhD
Nanotechnology Characterization Laboratory
Vinod Shah, PhD
Non Biological Complex Drugs Working Group
Jon de Vlieger, PhD
Lygature
Presented by

How to cite this eBriefing
The New York Academy of Sciences. Equivalence of Complex Drug Products: Scientific and Regulatory Challenges. Academy eBriefings. 2016. Available at: www.nyas.org/ComplexDrugs16-eB
Introduction
Speakers
Jon de Vlieger
Non Biological Complex Drugs Working Group
Scott McNeil
Lygature and Nanotechnology Characterization Laboratory
According to the Generic Pharmaceutical Association, almost 90% of prescriptions in the United States in 2015 were for generic drugs. Because generics are considered identical to their branded counterparts, pharmaceutical companies can forgo costly large-scale clinical trials. Instead, companies must demonstrate that a generic has pharmaceutical equivalence and bioequivalence to a licensed product. Pharmaceutical equivalence refers to the active ingredient, dosing, and strength, and can be confirmed via chemical and analytical methods. Bioequivalence refers to how a drug acts in the body and is evaluated via pharmacokinetic and/or pharmacodynamic studies. If these two conditions are met, regulatory agencies consider the generic to be safe and efficacious based on the clinical studies conducted on the branded drug.
Demonstrating the equivalence of generic complex drugs to their branded counterpart has unique challenges not encountered with small molecules.
Despite the success and advantages of generic drugs, users of certain drug categories, such as orally inhaled drugs used to treat asthma, currently have no access to generic counterparts. In addition, less than 50% of ophthalmic products and less than 40% of topical drugs have generic versions. That’s largely because these drugs represent complex drug products. In the newly released Generic Drug User Fee Act (GDUFA) “Regulatory Science Pritories,” the FDA defines complex drugs as those having complex active ingredients, such as peptides, polymers, or complex mixtures; complex formulations and/or dosages, such as liposomes, iron colloids, and long-acting injectables; complex routes of delivery, such as locally acting drugs, ophthalmic drugs, suspensions, emulsions, and gels; and/or complex drug–device combinations, such as nasal sprays and inhalers.
Demonstrating the equivalence of generic complex drugs to their branded counterpart has unique challenges not encountered with small molecules. First, the active components of complex drugs are often unknown, as are the critical attributes, i.e., the physical and chemical characteristics essential for efficacy and safety. Complex drugs are also difficult to fully characterize, and the products are very sensitive to changes in manufacturing, which is often proprietary.
In addition to above mentioned complex drug categories, the symposium also addressed biosimilars, generic versions of biologics, such as monoclonal antibodies, which share several of the hurdles to demonstrating equivalence.
Lawrence Mayer of Jazz Pharmaceuticals gave the keynote address on the development of a liposome-based drug delivery vehicle designed to maintain optimal levels of two cytostatics.
Several speakers spoke about the regulatory issues for biosimilars and complex drugs. Daan Crommelin of Utrecht University and Gillian Woollett of Avalere Health outlined some of the general issues to regulating generic complex drugs. Wenlei Jiang of the US Food and Drug Administration (FDA) and Elena Wolff-Holz of the Paul Ehrlich Institute provided the perspectives of the FDA and European Medicines Agency (EMA), respectively. Elywn Griffiths of the World Health Organization (WHO) outlined the WHO’s perspective.
There were several presentations on the approval of Glatopa (glatiramer acetate injection), a generic version of Copaxone (glatiramer acetate injection) for the treatment of multiple sclerosis. Andre Raw of the FDA outlined the evidence that the FDA required for approval of Glatopa while Joseph Glajch of Momenta Pharmaceuticals described the company’s approach to developing and characterizing it. Iris Grossman of Teva Pharmaceuticals presented data that show differences between the brand name drug and its generic counterpart.
Several generic versions of iron sucrose have been approved throughout the world. Stefan Mühlebach of Vifor Pharma presented evidence that several iron sucrose similars (ISS) are less effective than their branded counterparts. Beat Flühmann, also of Vifor, discussed his work on understanding pharmacists’ perspective on ISS.
Three speakers presented their approaches to evaluating the equivalence of complex drugs. Chetan Pujara of Allergan presented data on analyzing the equivalence of ophthalmic emulsions. Gary West of Azaya Therapeutics discussed regulatory hurdles to developing a generic form of Doxil (doxorubicin HCl liposome injection). Kouros Motamed of NantBioScience discussed the company’s efforts to develop a new drug delivery method for the cytostatic paclitaxel.
Two speakers focused on technical challenges to characterizing complex drugs. Wyatt Vreeland of the National Institute of Standards and Technology (NIST) described the advantages and caveats of several techniques to measure nanoparticle size. Stephan Stern of the Nanotechnology Characterization Laboratory (NCL) described a new technique to more accurately measure nanomedicines in the blood.
Speaker Presentation
Keynote Presentation
Keynote Speaker
Lawrence Mayer
Jazz Pharmaceuticals
Highlights
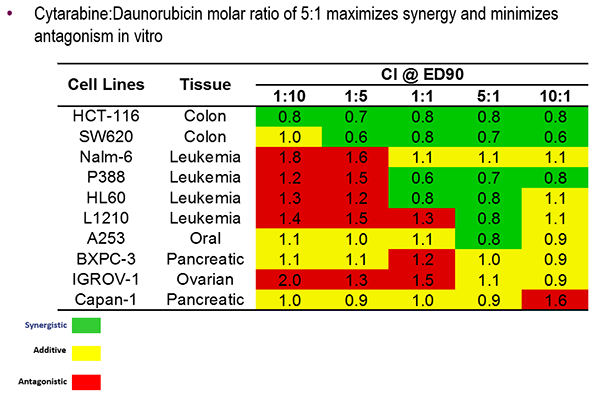
- Drug-delivery vehicles can control the pharmacokinetics of drug combinations to maintain a drug ratio that maximizes efficacy while minimizing toxicity.
- Vyxeos, a nanoscale drug delivery system, was developed to deliver two chemotherapies, daunorubicin and cytarabine, in a 5-to-1 molar ratio.
- In Phase 3 trials, Vyxeos demonstrated improved overall survival compared to daunorubicin plus cytarabine in patients with acute myeloid leukemia.
Built to scale
Lawrence Mayer described the development of Vyxeos (formerly CPX-351), a drug delivery system in Phase 3 trials for the treatment of acute myeloid leukemia (AML).
Vyxeos is a liposome-based drug delivery system that delivers the chemotherapies daunorubicin and cytarabine in a 5-to-1 ratio. In various cancer cell lines, this ratio was shown to have either additive or synergistic efficacy. Mayer described several of the technical features that can be altered to achieve the desired drug release rate, including membrane rigidity, charge, cholesterol content, and copper content.
Vyxeos was compared to the current chemotherapy regimen of cytarabine and daunorubicin in two Phase 2 and one Phase 3 clinical studies in patients with AML. In all three trials, patients treated with Vyxeos had a significantly higher overall survival. Other indicators of efficacy, such as response rates and post-transplant survival were also better in patients treated with Vyxeos. Jazz Pharmaceuticals is pursuing FDA approval of Vyxeos. The FDA granted the drug breakthrough status earlier this year.
Mayer stressed the importance of a scalable manufacturing process and biophysical characterization early in drug development. A well-characterized biophysical profile ensures that the product produced in large-scale production is the same as that produced and studied during small-scale testing. It also ensures that the features of the product do not vary considerably from batch to batch.
Speaker Presentation
00:00 Introduction
06:00 Key to exploiting drug ratio-dependent synergy
12:34 Leukemia cell update of intact liposomes
19:18 A robust commercial-scale nanomedicine
27:00 Conclusions
Regulatory Considerations
Speakers
Daan J.A. Crommelin, PhD
Utrecht University
Elwyn Griffiths
World Health Organization and Health Canada
Wenlei Jiang, PhD
Office of Research and Standards, Office of Generic Drugs, US Food and Drug Administration
Elena Wolff-Holz
Paul Ehrlich Institute
Gillian Woollett
Avalere Health
Panel Discussion Moderator:
Scott McNeil
Nanotechnology Characterization Laboratory
Highlights
- Regulatory pathways for small molecule generics and biosimilars are well established, while requirements for non-biologic complex drugs are considered on a case-by-case basis.
- The FDA is continually investigating new pharmacokinetic data and methods to assess the bioequivalence of complex drugs.
- New guidance was released by the EMA in 2014 that expands the definition of biosimilar and provides additional direction to industry.
Regulatory pathways for generics and biosimilars
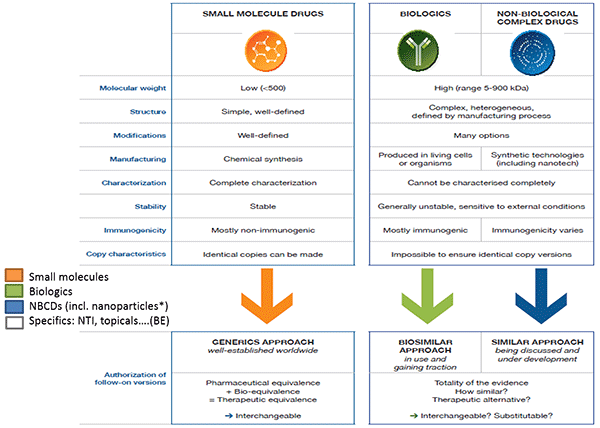
Daan Crommelin began by defining some key terms relating to complex drug products and outlining the regulatory pathways for different types of drugs. According to Crommelin, drugs can be divided into three main categories: small molecules; biologics, such as vaccines, blood and blood components, gene therapies, tissues, and recombinant proteins; and non-biologic complex drugs (NBCDs), synthetic drugs that do not have a single, homogeneous active substance and cannot be fully characterized. The composition, quality and in vivo performance of NBCDs are highly dependent on the manufacturing processes of the active ingredient as well as (in most cases) the formulation. They often consist of nanomedicines such as polymeric micelles, liposomes, or peptide-polymers. Crommelin pointed out that regulatory agencies do not always agree on these definitions; while the EMA considers low molecular weight heparin a biologic, the FDA considers it a complex drug.
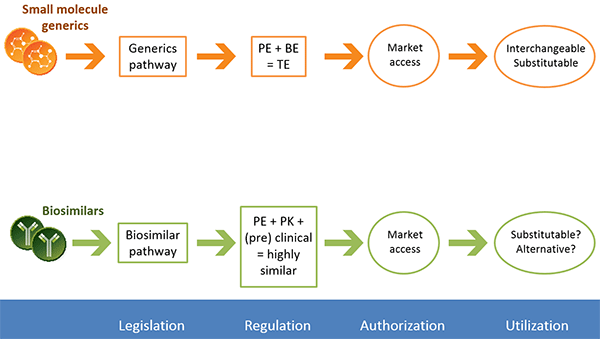
The regulatory approval process for generic small molecules is well established, and generics are considered interchangeable with their branded counterpart. A separate process has been created for biologics. Similar to generics, biosimilars can be approved via an abbreviated regulatory process that usually involves pharmaceutical equivalence, pharmacokinetic, and preclinical data. Biosimilars are considered highly similar, but not identical, to the original drug.
The regulatory process for NBCDs is often conducted on a case-by-case basis. Because NBCDs cannot be fully characterized and the critical attributes required for efficacy are often unknown, it is difficult to demonstrate bioequivalence for a generic NBCD. The FDA and EMA release guidance documents for NBCD classes outlining which analyses are needed to demonstrate similarity. These documents are continually reviewed and revised.
Multi-faceted considerations for regulatory approval of biosimilars and complex drugs
Gillian Woollett stressed that the considerations for biosimilars and complex drug similars are not only scientific, but also legal, regulatory, and economic. Regulatory agencies must balance the need for scientific and clinical evidence with the need to provide patients affordable drugs in a timely manner. According to Woollett, too high a standard can be as problematic as too low a standard if it prevents access to drugs. The FDA and EMA have an additional responsibility to set a standard for the rest of the world. A product approved in the EU and US can generally be approved anywhere; however, one approved in emerging markets, such as China, Russia, and Brazil, may not be approved in other markets. In the US, only four biosimilars have been approved; however, several agents have been approved under the definition of generic biologics. In comparison, 23 biosimilars have been approved in the EU.
The considerations for biosimilars and complex drug similars are not only scientific, but also legal, regulatory, and economic.
One issue surrounding biosimilars is that they must be compared to the reference biologic approved in a given market. This can make it difficult for companies to pursue approval globally. Another issue that may need to be addressed is the addition of orphan indications to reference biologics. Because the FDA gives exclusivity for orphan indications, these indications would not be included in the biosimilar’s product label.
The FDA’s perspective on determining the equivalence of complex drugs
Wenlei Jiang described how the FDA assesses the bioequivalence of complex generics. Jiang stressed the FDA’s commitment to complex generics through the abbreviated new drug application (ANDA) approval pathway and GDUFA, which employs regulatory science initiatives for generic drugs. Specifically, Jiang showed research efforts under GDUFA that have developed new bioequivalence methods for complex drugs.
Traditional pharmacokinetic studies to demonstrate the bioequivalence of complex drugs are difficult to conduct. For some drugs, such as long-acting polymeric microspheres, which are commonly used in drug delivery, their long-acting nature would require very long studies, which may be impractical. Based on pharmacokinetic modeling research conducted by Alkermes, the FDA updated its guidance on the bioequivalence requirements for long-acting polymeric microspheres to include drug exposure up to 28 hours.
For complex drugs that act locally, such as topical skin products and nasal sprays, conventional studies that monitor drug blood levels may not be relevant. The FDA has sponsored research on new options for dermal pharmacokinetic studies, such as open-flow microperfusion (see slide).
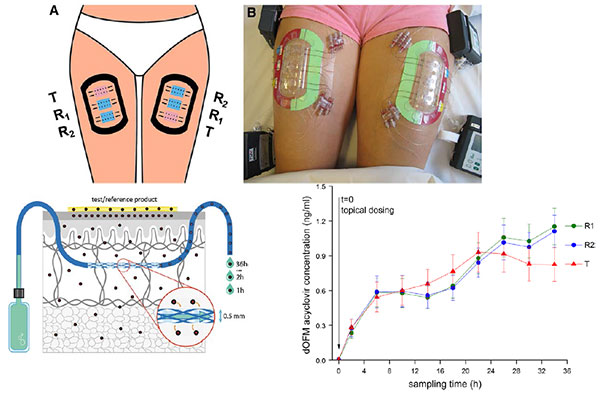
Jiang also presented a new analytic technique, morphology-directed Raman spectroscopy (MDRS), that can determine drug particle size and shape within a nasal suspension. MDRS has supported the recent approval of mometasone nasal suspension.
CHMP/EMA perspective on biosimilars
Elena Wolff-Holz described the EMA’s perspective on evaluating biosimilars and how the process has evolved over time. The EMA releases several overarching and product-specific guidelines, which are continually reviewed and revised. In a recent update to the overarching guidance (CHMP/437/04 Rev. 1), the EMA expanded the concept of a biosimilar to any biological medicinal product, including vaccines, allergens, and cell and tissue therapies, which were previously excluded. It also stressed that a biosimilar contains “a version of the active substance of an already authorized original biological medicinal product,” implying that biosimilars should not be used as reference products. In the new guidance, the goal of clinical data is to address slight differences in product characteristics, and safety and efficacy do not need to be re-established. Finally, if the reference biologic receives a new indication, the indication of the biosimilar may also be extended without the need for additional studies, if scientifically justified.
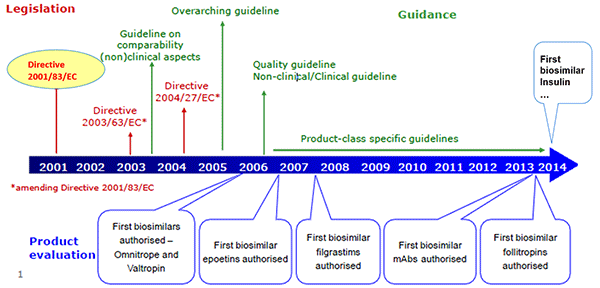
Wolff-Holz noted that the approval process for biosimilars, which emphasizes physicochemical properties, can be difficult for clinicians, who want to know the clinical information for a drug, to accept. While clinical data for biosimilars are not included in the product’s Summary of Product Characteristics (SmPC), they are in the European Public Assessment Reports (EPAR), which clinicians are not as familiar with.
The WHO’s role in biosimilars and complex generics
Elwyn Griffiths described WHO’s role and position on biosimilars. While WHO is not a regulatory agency, it sets global standards for safety and quality. The WHO Expert Committee on Biological Standardization meets annually to establish recommendations and guidance on the quality, safety, and efficacy of biologics.
There are three major WHO guidance documents pertaining to biologics and biosimilars: Guidelines on the quality, safety and efficacy of biotherapeutic protein products prepared by recombinant DNA technology, published in 2013, Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks, published in 2010, and Guidelines on the evaluation of similar biotherapeutic products, published in 2009.
The 2009 guidelines on similar biotherapeutics define similar biotherapeutic products (SBPs) as similar in terms of quality, safety, and efficacy to a licensed reference biological product (RBP). Similarity is established by characterizing the SBP and comparing it to the RBP in head-to-head studies. Comparison trials should be designed to detect differences in safety and efficacy between the SBP and RBP, not to establish the efficacy of the SBP. The guidelines also state that if an RBP has more than one indication, the indication of the SBP may also be expanded if scientifically warranted. One subject the WHO guidelines leave up to national regulatory agencies is whether SBPs should be considered interchangeable with RBPs.
Speaker Presentations
00:00 Introduction
01:52 Complex drug products definitions
07:47 Three drug classes
13:16 What is the issue?
17:47 No access to biosimilar pathway for NBCD-similars
00:00 Introduction
03:35 WHO biological standards
09:29 Regulatory oversight of biotherapeutics
15:13 WHO guidelines on evaluation of similar biotherapeutic product
23:38 Summary WHO standards
00:00 Introduction
02:32 NDA versus ANDA
07:12 Consideration on PLGA sameness
13:24 Challenges for therapeutic equivalence of nasal spray products
15:13 Scientific rationale for weight-of-evidence approach
20:54 Summary
00:00 Introduction
03:03 Scope includes next generation biosimilars
10:43 Manufacturing changes authorized by EMA
18:09 What if PK similarity is not convincingly demonstrated?
00:00 Introduction
01:38 Working definitions for biologics
05:51 Biologic approvals have increased and are expected to rise
10:27 Biosimilars approvals across the world
18:58 Biosimilars represent a new and different business
00:00 Introduction
00:50 Thoughts on the definition of the API and does it need to be updated
05:48 Measuring release of drugs
07:40 Critical factors, quality attributes for complex drugs
17:51 Looking forward to how further complexity affects biological responses
21:49 IP and open data review in a profit-driven industry
Comparisons of Copaxone (glatiramer acetate injection) with Its Generic Glatopa (glatiramer acetate injection)
Speakers
Joseph Glajch
Momenta Pharmaceuticals
Iris Grossman
Teva Pharmaceuticals
Andre Raw
U.S. Food and Drug Administration
Highlights
- Glatopa (glatiramer acetate injection), an FDA-approved generic of Copaxone(glatiramer acetate injection) is considered similar to the branded product based on the tests required by the FDA.
- Additional comparisons have revealed differences between the drugs that may affect safety and efficacy.
Glatopa: a complex generic drug for multiple sclerosis
Glatopa was approved by the FDA in 1996 as a generic version of Copaxone for the treatment of multiple sclerosis. These drugs consist of glatiramer acetate, a complex mixture of polypeptides produced by polymerizing four amino acids and then partially depolymerizing the resulting peptides.
Scientific considerations to approaching complex generics
Andre Raw discussed the scientific considerations to approaching generic versions of Copaxone (glatiramer acetate) and Lovenox (enoxaparin sodium) injections, two of the complex active ingredients recently approved in generic versions by the FDA. Raw argued that understanding the nature of the complexity of these drugs is key to evaluating generic similars. For instance, the complexity of Copaxone comes from the polymerization of activated amino acids, which determines the sequences of the polypeptides, and the subsequent partial depolymerization.
To ensure similarity of Copaxone and Lovenox similars, the FDA drafted guidance listing four criteria that generic products must meet. First, the pharmaceutical company must demonstrate equivalence in its reaction scheme. Second, the FDA places constraints on several physicochemical properties, such as molecular weight distribution and amino acid composition. This also means that the structural signatures, which reflect the polymerization and depolymerization processes, and biological assays will be similar as well.
The third criterion requires that generic manufacturers identify “process signatures;” equivalence in these signatures ensures that the generic active ingredient is manufactured with effectively the same polymerization kinetic and cleavage biases as the brand name. Finally, the fourth criterion relies on the equivalence of biochemical and/or biological markers for activity to ensure sameness. Unlike the previous three criteria, which in their aggregate ensure equivalency of molecular diversity, this criterion serves as a confirmatory test to ensure equivalency in functional biochemical or biological markers.
Comparison of Copaxone and Glatopa for FDA approval
Joseph Glajch described the tests that Sandoz conducted to demonstrate the equivalence of Glatopa and Copaxone. In approximately 50 physiochemical and 10 biological tests, Glatopa fell within an acceptable range considered to be equivalent to Copaxone. Glajch stressed that these analyses must be conducted on dozens of lots to understand the diversity and variability within the drug. He showed the data from some of these analyses, including molecular weight distribution, amino acid composition, and structural signatures. He also showed that the drugs have similar effects on disease intensity in a model of autoimmune encephalomyelitis and similar results on histamine release from cells.
Additional comparisons of Copaxone and Glatopa
While Raw and Glajch showed that Glatopa and Copaxone are similar according to the methods required by the FDA, Iris Grossman described studies using higher-resolution techniques conducted by Teva Pharmaceuticals that reveal differences in surface charge and molecular weight density and distribution. In addition, Glatopa had greater batch-to-batch variability in these characteristics than Copaxone.
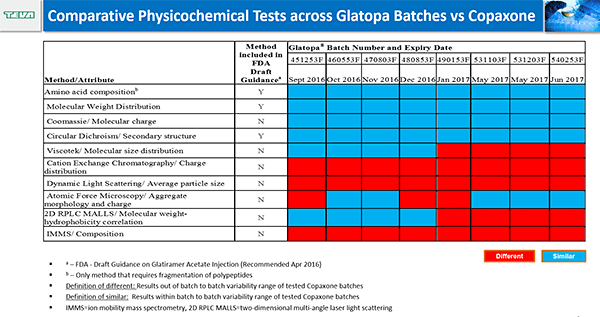
These differences may have consequences on efficacy and safety. In mice, of the thousands of genes affected by Glatopa and Copaxone, approximately 10% were different between the two drugs. The genes that differed were involved in pathways important for efficacy, including inflammation and immune response.
Based on these results, Teva has submitted comments on the draft FDA guidance on glatiramer acetate injections.
Speaker Presentation
00:00 Introduction
02:37 Physicochemical differences relevant to mode of action
10:22 Relevance of differences to function and safety
13:50 Immunological triad
17:25 Functional effects summary and conclusions
Issues with Iron Carbohydrate Similars
Speakers
Beat Flühmann
Vifor Pharma Ltd
Stefan Mühlebach, PhD
Vifor Pharma Ltd
Panel Discussion Moderator:
Daan J.A. Crommelin
Utrecht Universit
Highlights
- Several approved generics of iron carbohydrate complexes are not as effective as their branded counterparts.
- Hospital pharmacists are not aware of the differences between iron carbohydrate products and often substitute these products without informing a patient’s physician.
Iron carbohydrate similars
Iron carbohydrate products are used to treat anemia in patients with chronic kidney disease. Several iron carbohydrate generics were approved in Europe in the mid-2000s. According to Stefan Mühlebach, there was no regulatory process for complex similars at this time, and the drugs were approved via the small molecule generic route. Researchers and regulators were not aware that the active pharmaceutical ingredient was not fully characterized, and iron carbohydrate similars were deemed identical to and interchangeable with the original drugs without additional efficacy or safety studies. Since then, real-life clinical studies have shown that show the approved iron carbohydrate similars are not clinically identical to the original formulations.
The EMA and FDA have reacted to these data and have published several guidance papers on iron carbohydrate similars.
The hospital pharmacist’s approach to iron complex carbohydrates
Beat Flühmann of Vifor Pharma described the pharmacist’s perspective of complex generics using iron sucrose, a type of iron carbohydrate, as an example. In a survey hospital pharmacists in France and Spain, Flühmann found that pharmacists substitute 30% to 50% of branded iron sucrose prescriptions with a generic version. Approximately 40% of pharmacists believed that the products have the same efficacy and safety, while the remaining 60% had no opinion. In addition, many pharmacists did not inform the physician that they had dispensed the generic version.
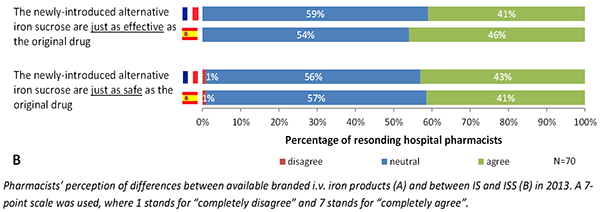
Flühmann stressed the importance of educating pharmacists on the differences in biosimilars and complex generics. Vifor conducted a roundtable discussion earlier this year with leading hospital pharmacists throughout Europe to discuss how to select nanosimilars for hospital formularies. The roundtable added several criteria for hospital formulary committees to consider when selecting a nanosimilar, including particle size and size distribution, pharmacokinetics, and stability of ready-to-use formulations.
Speaker Presentations
00:00 Introduction
04:40 Therapeutic equivalence from manufacturing to efficacy and safety
09:32 Current situation for follow-on approval
14:48 NBCD product evaluation
18:32 Landscape of drugs follow-ons
00:00 Introduction
03:11 Making biosimilars available globally
08:25 The unbound drug vs release and release mechanisms
12:11 Interchangeability and acceptability in the market
16:27 Proposed systems for biosimilars and for currently approved biologics
20:12 Concluding thoughts
Other Case Studies of Complex Generics
Speakers
Kouros Motamed
NantBioScience, Inc.
Chetan Pujara
Allergan
Gary West
Azaya Therapeutics
Highlights
- The bioequivalence of Cynviloq, a polymeric micelle formulation of the chemotherapy paclitaxel, and Abraxane (paclitaxel protein-bound particles for injectable suspension), an approved albumin-bound paclitaxel, is being investigated in the study TRIBECA.
- Robust criteria are needed to ensure the equivalence of different ophthalmic emulsions.
- Greater harmony between the FDA and EMA could streamline the approval of a generic version of Doxil (doxorubicin HCl liposome injection).
Cynviloq: a new formulation of paclitaxel
Kouros Motamed discussed a polymeric micelle formulation of the chemotherapy paclitaxel, Cynviloq, which is approved in several Asian countries for breast and lung cancer under the brand name Genexol-PM. These approvals were based on comparisons to Taxol (paclitaxel), a formulation of paclitaxel containing the excipient Cremophor EL. In the US and EU, approval requires comparison to Abraxane, an albumin-bound formulation of paclitaxel approved for breast, lung, and pancreatic cancer.
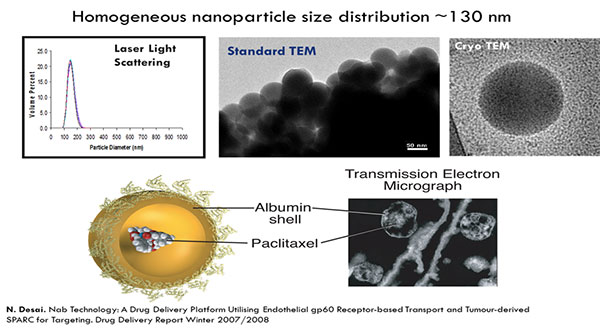
In in vitro studies, Abraxane and Cynviloq had similar pharmacokinetic properties at therapeutic doses. At higher doses, the Abraxane concentration sharply increased, resulting in increased toxicity. The concentration of Cynviloq, however, did not increase as quickly, and therefore may be less toxic at higher doses.
A pharmacokinetic bioequivalence study, TRIBECA, was conducted in patients with metastatic breast cancer. Sorrento Therapeutics issued a press release stating that TRIBECA demonstrated bioequivalence of Abraxane and Cynviloq. In 2015, Cynviloq was acquired by NantWorks and has been renamed Nant-paclitaxel.
Demonstrating equivalence of ophthalmic emulsions
In 2013, the FDA released draft guidance on cyclosporine emulsions. To test whether this guidance was sufficient, Allergan tweaked its manufacturing process to make nine cyclosporine emulsions similar to Restasis (cyclosporine ophthalmic emulsion). Chetan Pujara showed that although these emulsions met the FDA criteria for similarity, there were key physicochemical and biological differences. For example, the cyclosporine distribution between the phases of the emulsion varied between the drugs. This corresponded with differences in the drug’s ability to permeate human corneal cells. Allergan also saw variability in the amount of cyclosporine in tears in rabbits.
The updated FDA guidance, released in 2016, incorporates suggestions by Allergan based on these data. However, Pujara stressed that further research on linking in vitro results to in vivo performance and on robust methods to characterize emulsions is still needed.
Regulatory barriers to a generic version of Doxil
Gary West called for harmony between regulatory authorities, citing the recent difficulties that Azaya Therapeutics has had in gaining approval for its generic doxorubicin. Doxorubicin, a treatment for ovarian cancer, is marketed by Janssen in the US under the brand name Doxil and in the EU under the brand name Caelyx (pegylated liposomal doxorubicin hydrochloride for injection).
Can international regulatory agencies work together to remove barriers facing approval of generic drugs?
Currently, Caelyx is the reference drug for doxorubicin in the EU. In the US, the FDA established a generic version of doxorubicin, ANDA 203263, as the reference drug for doxorubicin due manufacturing concerns with Doxil. However, the FDA discovered that the plant manufacturing the generic version was not complying with several guidelines, which led to contamination. West argued that due to these issues, the FDA-approved doxorubicin generic may not be the best comparator. He showed certificates of analysis from the sole manufacturer that made both Caelyx and Doxil in 2013 that demonstrate that, at that time, the two drugs were the same product with a different label and argued that they should therefore be considered interchangeable by regulatory agencies.
Azaya Therapeutics’ investigational doxorubicin generic, dubbed ATI-0918, was shown to be bioequivalent to Caelyx. However, because the EMA requires that Caelyx be sourced within the EU, and the FDA requires generics to be compared against the reference standard ANDA 203263, the company faced regulatory hurdles from both agencies. West called upon the agencies to work together to remove the barriers facing approval of generic drugs.
Speaker Presentation
00:00 Introduction
03:21 A triple-down, unintended, regulatory catch 22
09:52 Lipodox versus Sun product
18:28 Regulatory barriers
22:29 Conclusions
Analytical Techniques for Characterizing Complex Drugs
Speakers
Stephan Stern
Nanotechnology Characterization Laboratory
Wyatt Vreeland
The National Institute of Standards and Technology
Highlights
- Choosing the right analytical method is important when measuring the size of nanoparticles.
- A new method to measure plasma drug concentration for nanomedicines is being validated by the NCL.
Techniques to measure nanoparticle size
Wyatt Vreeland described the advantages and disadvantages of several techniques to measure nanoparticles size.
The most common technique for measuring nanoparticle size is batch light scattering. In this method, the particle size is determined based on their ability to scatter light waves. This method is quick, simple, and nondestructive. However, because larger particles scatter light much more than smaller particles, a small amount of large particles can drown out the signal due to smaller particles, resulting in significant errors.
To avoid this complication, mixtures can be separated by size by either size-exclusion chromatography (SEC) or field flow fractionation (FFF) and then measured by light scattering. One advantage of FFF over SEC is that it can separate delicate or soft colloidal particles, which are often components of complex drugs. FFF can be used to separate particles of a few nanometers to 1000 nanometers.
Vreeland also discussed several new techniques that measure particle size by monitoring individual particles. In nanoparticle tracking analysis (NTA), particles are imaged directly with a laser, and their size is determined by tracking the extent of diffusion over time. While NTA is a robust tool to measure particle size, it is only effective within a narrow concentration range. A second technique, resonant mass measurement measures the change in frequency a particle causes as it flows through a vibrating cantilever. In addition to size, this technique, unlike others, can also measure buoyant density; however the cantilever is prone to clogging and cannot be used to measure liposomes. Finally, electrical sensing zone measures the change in electrical conductivity as a particle travels through a pore between two electrodes. This technique is similar to Coulter counter, which is used to measure bacteria and viruses. This is a very new technique for nanoscale particles.
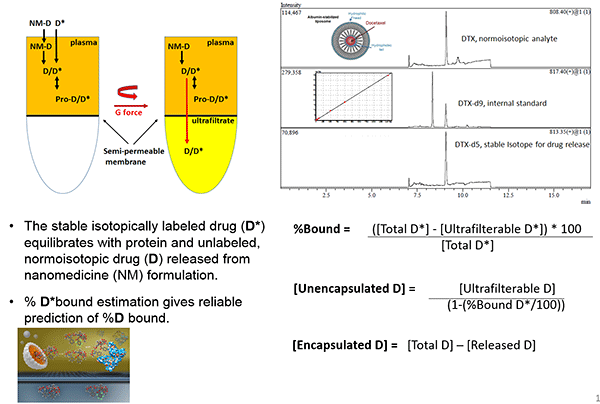
A new method to measure nanomedicine plasma levels
The concentration of drugs administered via nanoparticles can be divided into three forms: the nanoparticle-encapsulated form, the unencapsulated free form, and the unencapsulated protein-bound form. Current methods to separate the encapsulated from unencapsulated drug promote un-encapsulation and do not differentiate protein-bound from encapsulated drug.
In two pharmacokinetic studies of Caelyx, the measured levels of unencapsulated drug differed by 6-fold, while the levels of unencapsulated drug differed by almost 12-fold, likely due to problems in fractionation methods.
Stephan Stern presented a new stable isotope tracer technique being developed by the NCL to separate and measure nanomedicine drug fractions. NCL is working with the FDA to evaluate this method for determining the bioequivalence of generic nanomedicines. As part of this effort, the NCL is conducting in vitro bioanalytical studies as well as bioequivalence studies in rats of Abraxane and Genexol-PM, discussed by Motamed, and doxorubicin HCl and Doxil, discussed by West.
Speaker Presentations
00:00 Introduction
03:48 Nanomedicine pharmacokinetics
08:39 Sources of intra-subject variability
13:43 Novel stable isotope tracer method
19:26 Measuring unbound drug in evaluating nanomedicine pharmacokinetics
00:00 Introduction
02:50 Batch light scattering
08:11 How laminar flow works in FFF
13:18 Dynamic light scattering
17:52 Resonant mass measurement
20:18 Future directions
Open Questions
- How can researchers determine the critical attributes for complex drugs?
- What are the best methods to characterize complex drugs?
- What are the most appropriate pharmacokinetic methods to evaluate bioequivalence of complex drugs?
- Should biosimilars and complex generics be considered interchangeable with their branded counterparts? What criteria are necessary to demonstrate interchangeability?
- How can regulatory agencies balance the need for robust scientific and clinical evidence with the need for affordable medications when evaluating complex generics?
- How can regulatory agencies reach agreements on how to evaluate complex drugs?
- How can companies and regulatory agencies communicate the potential clinical differences between original and generic complex drugs?
Resources
Websites
Generic Drug User Fees Act (GDUFA) of 2012
Information from the FDA on GDUFA 2012 as well as a webinar held in October, 2016 on GDUFA II
GDUFA II Fee Structure Summary
Summary of changes under GDUFA II
GDUFA regulatory science
News and information on GDUFA from the FDA
Generic Pharmaceutical Association
Trade association for manufacturers and distributors of generic drugs
Generics and Biosimilars Initiative
Initiative to promote the scientific status of generics and biosimilars to foster the use of affordable medicines
WHO biologics site
Biological standardization website
Daan Crommelin
Borchard G, Fluhmann B, Muhlebach S. Nanoparticle iron medicinal products — Requirements for approval of intended copies of non-biological complex drugs (NBCD) and the importance of clinical comparative studies. Regul Toxicol Pharmacol. 2012;64:324-328.
Crommelin DJ, de Vlieger JS, Weinstein V, Muhlebach S, Shah VP, Schellekens H. Different pharmaceutical products need similar terminology. AAPS J. 2014;16:11-14.
Crommelin DJ, Shah VP, Klebovich I, et al. The similarity question for biologicals and non-biological complex drugs. Eur J Pharm Sci. 2015;76:10-17.
Ebbers HC, Crow SA, Vulto AG, Schellekens H. Interchangeability, immunogenicity and biosimilars. Nat Biotechnol. 2012;30:1186-1190.
Halim LA, Brinks V, Jiskoot W, et al. Quality and batch-to-batch consistency of original and biosimilar Epoetin products. J Pharm Sci. 2016;105:542-550.
Praditpornsilpa K, Tiranathanagul K, Kupatawintu P, et al. Biosimilar recombinant human erythropoietin induces the production of neutralizing antibodies. Kidney Int. 2011;80:88-92.
Beat Flühmann
Fluhmann B, de Vlieger JSB, Vulta AG, et al. The authorization of non-biological complex drugs (NBCDs) follow-on versions: specific regulatory and interchangeability rules ahead? GaBI J. 2013;2:204-207.
Joseph Glajch
FDA draft guidance document on glatiramer acetate injection
Elwyn Griffiths
WHO Expert Committee on Biological Standardization
WHO. Guidelines on the evaluation of similar biotherapeutic products. 2009.
Case studies and publications from implementation workshops for BTP/SBP guidelines:
Evaluation of similar biotherapeutic products: scientific and regulatory challenges. Biologicals [special issue]. 2011;39:5.
Knezevic I, Kang HN, Thorpe R. Immunogenicity assessment of monoclonal antibody products: A simulated case study correlating antibody induction with clinical outcomes. Biologicals. 2015;43:307-317.
Kudrin A, Knezevic I, Joung J, Kang HN. Case studies on clinical evaluation of biosimilar monoclonal antibody: scientific considerations for regulatory approval. Biologicals. 2015;43:1-10.
Njue C. Statistical considerations for confirmatory clinical trials for similar biotherapeutic products. Biologicals. 2011;39:266-269.
Schiestl M, Li J, Abas A, et al. The role of the quality assessment in the determination of overall biosimilarity: a simulated case study exercise. Biologicals. 2014;42:128-132.
Wadhwa M, Knezevic I, Kang HN, Thorpe R. Immunogenicity assessment of biotherapeutic products: an overview of assays and their utility. Biologicals. 2015;43:298-306.
Iris Grossman
Hasson T, Kolitz S, Towfic F, et al. Functional effects of the antigen glatiramer acetate are complex and tightly associated with its composition. J Neuroimmunol. 2016;290:84-95.
Kolitz S, Hasson T, Towfic F, et al. Gene expression studies of a human monocyte cell line identify dissimilarities between differently manufactured glatiramoids. Sci Rep. 2015;5:10191.
Varkony H, Weinstein V, Klinger E, et al. The glatiramoid class of immunomodulator drugs. Expert Opin Pharmacother. 2009;10:657-668.
Teva Pharmaceuticals Industries Ltd. [press release] Teva Files Citizen Petition with the U.S. Food and Drug Administration (FDA) Regarding the Complexity of COPAXONE® (glatiramer acetate) Following the Agency’s Guidance. July 3, 2014.
Wenlei Jiang
Babiskin A et al. Pharmacokinetic modeling and simulation of naltrexone for extended-release injectable suspension to derive alternative BE metrics. ACoP. 2015.
Bodenlenz M, Tiffner KI, Raml R, et al. Open flow microperfusion as a dermal pharmacokinetic approach to evaluate topical bioequivalence. Clin Pharmacokinet. 2016. Epub ahead of print.
Garner J, Skidmore S, Park H, Park K, Choi S, Wang Y. A protocol for assay of poly(lactide-co-glycolide) in clinical products. Int J Pharm. 2015;495:87-92.
Li BV, Jin F, Lee SL, et al. Bioequivalence for locally acting nasal spray and nasal aerosol products: standard development and generic approval. AAPS J. 2013;15:875-883.
Lawrence Mayer
Phase 3 clinical study of Vyxeos in AML
Carol H, Fan MM, Harasym TO, et al. Efficacy of CPX-351, (cytarabine:daunorubicin) liposome injection, against acute lymphoblastic leukemia (ALL) xenograft models of the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2015;62:65-71.
Cortes JE, Goldberg SL, Feldman EJ, et al. Phase II, multicenter, randomized trial of CPX-351 (cytarabine:daunorubicin) liposome injection versus intensive salvage therapy in adults with first relapse AML. Cancer. 2015;121:234-242.
Lancet JE, Cortes JE, Hogge DE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014;123:3239-3246.
Lim WA, Tardi PG, Dos Santos N, et al. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytarabine: daunorubicin formulation, in bone marrow xenografts. Leuk Res. 2010;34:1214-1223.
Kouros Motamed
Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41:2971-3010.
Motamed K, Goodman Y, Hwang L, Hsiao C, Trieu V. IG-001 — a non-biologic nanoparticle paclitaxel for the treatment of solid tumors. J Nanomater Mol Nanotechnol. 2014;3:1.
Nehate C, Jain S, Saneja A, et al. Paclitaxel formulations: challenges and novel delivery options. Curr Drug Deliv. 2014;11:666-686.
Sorrento Therapeutics, Inc. Sorrento Announces Positive TRIBECA™ Registrational Study Results. [press release.] May 4, 2015.
Stefan Mühlebach
Aguera ML, Martin-Malo A, Alvarez-Lara MA, et al. Efficiency of original versus generic intravenous iron formulations in patients on haemodialysis. PLoS One. 2015;10:e0135967.
Muhlebach S, Borchard G, Yildiz S. Regulatory challenges and approached to characterize nanomedicines and their follow-on similars. Nanomedicine. 2015;10:659-674.
Munoz M, Martin-Montanez E. Ferric carboxymaltose for the treatment of iron-deficiency anemia. Exp Opin Pharmacother. 2012;13:907-921.
Toblli JE, Cao G, Angerosa M. Nitrosative stress and apoptosis in non-anemic healthy rats induced by intravenous iron sucrose similars versus iron sucrose originator. Biometals. 2015;28:279-292.
Toblli JE, Di Gennaro F. Switching patients with non-dialysis chronic kidney disease from oral iron to intravenous ferric carboxymaltose: effects on erythropoiesis-stimulating agent requirements, costs, hemoglobin and iron status. PLoS One. 2015;10:e0125528.
Chetan Pujara
FDA. Draft guidance on cyclosporine. Revised October, 2016.
Kompella Uday. Approval of Generic Ophthalmic Suspensions/Emulsions: Current Status and Challenges. Regulatory Science Initiatives Part 15 Public Meeting.
Lionberger R. Complex Generic Drugs. GPhA Fall Technical Meeting, October 29, 2013.
Andre Raw
Department of Health and Human Services. FDA Letter to Covington and Burling (Docket FDA-2003-P-0273). July 23, 2010.
Department of Health and Human Services. Requests That FDA Consider New Scientific Information and Refrain From Approving Any Abbreviated New Drug Application Referencing Copaxone (glatiramer acetate injection) Until Certain Conditions Are Met (Docket FDA-2015-P-1050). April 16, 2015.
Grampp G, Bonafede M, Felix T, Li E, Malecki M, Sprafka JM. Active and passive surveillance of enoxaparin generics: a case study relevant to biosimilars. Expert Opin Drug Saf. 2015;14:349-360.
Lee S, Raw A, Yu L, et al. Scientific considerations in the review and approval of generic enoxaparin in the United States. Nat Biotechnol. 2013;31:220-226.
Stefan Stern
Ambardekar and Stern. NBCD Pharmacokinetics and Bioanalytical Methods to Measure Drug Release. In Daan Crommelin D and de Vlieger J (ed) Non-Biological Complex Drugs; the Science and Regulatory Landscape. Springer, New York, NY; 2015: In-press.
Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ. Plasma protein binding of amphotericin B and pharmacokinetics of bound versus unbound amphotericin B after administration of intravenous liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate. Antimicrob Agents Chemother. 2002;46:834-840.
Davit BM, Conner DP, Fabian-Fritsch B, et al. Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPS J. 2008;10:148-156.
Skoczen S, McNeil SE, Stern ST. Stable isotope method to measure drug release from nanomedicines. J Control Release. 2015;220:169-174.
Stern ST, Martinez MN, Stevens DM. When is it important to measure unbound drug in evaluating nanomedicine pharmacokinetics? Drug Metab Dispos. 2016;44:1934-1939.
Wyatt Vreeland
Galyean AA, Filliben JJ, Holbrook RD, Vreeland WN, Weinberg HS. Asymmetric flow field flow fractionation with light scattering detection — an orthogonal sensitivity analysis. J Chromatogr A. 2016;1473:122-132.
Galyean AA, Vreeland WN, Filliben JJ, Holbrook RD, Ripple DC, Weinberg HS. Using light scattering to evaluate the separation of polydisperse nanopartciles. Anal Chim Acta. 2015;886:207-213.
Gary West
Zerhouni E, Hamburg M. The need for global regulatory harmonization: a public health imperative. Sci Transl Med. 2016;8:338ed6.
Elena Wolff-Holz
EMA. Guideline on similar biological medicinal products. CHMP/437/04 Rev 1. 23 October 2014.
McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91:405-417.
Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28:363-372.
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nature Biotechnol. 2011;29:310-312.
Vezer B, Buzas A, Sebeszta M, Zrubka Z. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin. 2016;32:829-834.
Gillian Woollett
McCamish M, Woollett, G. Worldwide experience with biosimilar development. MAbs. 2011;3:209-217.
McCamish, M, Pakulski, J, Sattler C, Woollett G. Toward interchangeable biologics. Clin Pharmacol Ther. 2015;97:215-217.
Biosimilar guidances:
CHMP. Guideline on similar biological medicinal products. CHMP/437/04. 30 October 2005.
FDA. Information on biosimilars.
Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs). March 2010.
WHO. Guidelines on evaluation of similar biotherapeutic products (SBPs). October 2009.