Linking Metabolism, Health, and Cancer: 2020 Dr. Paul Janssen Award Symposium
Reported by:
Alla Katsnelson
Presented by:
The Dr. Paul Janssen Award for Biomedical Research
The New York Academy of Sciences
Lewis C. Cantley’s discovery of the enzyme phosphatidylinositol-3-kinase (PI3K) paved the way for a better understanding of cellular metabolism and its role in human diseases. In response to insulin, PI3K signals through lipids to activate a cellular cascade resulting in increased glucose uptake and subsequent cell growth and division. Cantley’s work has led to new cancer therapies, as PI3K pathway mutations are among the most common to drive cancer development, and a better understanding of insulin resistance in diabetes.
For his groundbreaking discovery of PI3K and critical body of research, Lewis C. Cantley, PhD, of Weill Cornell Medical College, received the 2020 Dr. Paul Janssen Award for Biomedical Research. On September 16, 2020, the New York Academy of Sciences hosted the award symposium to celebrate his achievements. Following Cantley’s award lecture, other experts in the field shared their work on the intersection between cellular metabolism, biology and disease.
Symposium Highlights
- Differences in cell metabolism across cancer types may help explain why cancer cells are differentially sensitive to drugs that target metabolism.
- The tumor suppressor protein p53 regulates several pathways that manage the production of reactive oxygen species in the mitochondria.
- Metabolic pathways represent a powerful and to-date, underappreciated set of therapeutic targets for cancer.
- Drugs that regulate metabolic pathways involved in cell differentiation are promising targets for acute myeloid leukemia.
- Cancer researchers are coming to appreciate how a patient’s diet can be therapeutically applied and may directly modulate their disease progression and therapeutic response.
Speakers
Lewis C. Cantley, PhD
Weill Cornell Medical College
Matthew Vander Heiden, PhD
Massachusetts Institute of Technology
Karen Vousden, PhD
Francis Crick Institute
Ulrike Philippar, PhD
Johnson & Johnson
Costas Lyssiotis, PhD
University of Michigan
Discovering Phosphatidylinositol-3-kinase and Its Link to Cancer
Speakers
Lewis C. Cantley
Weill Cornell Medical College
Metabolism, Health, and Cancer
Lewis C. Cantley described how the discovery of the enzyme phosphatidylinositol-3-kinase (PI3K) and its signaling pathway led to the development of a new class of cancer drugs called phosphoinositide-3 kinase inhibitors.
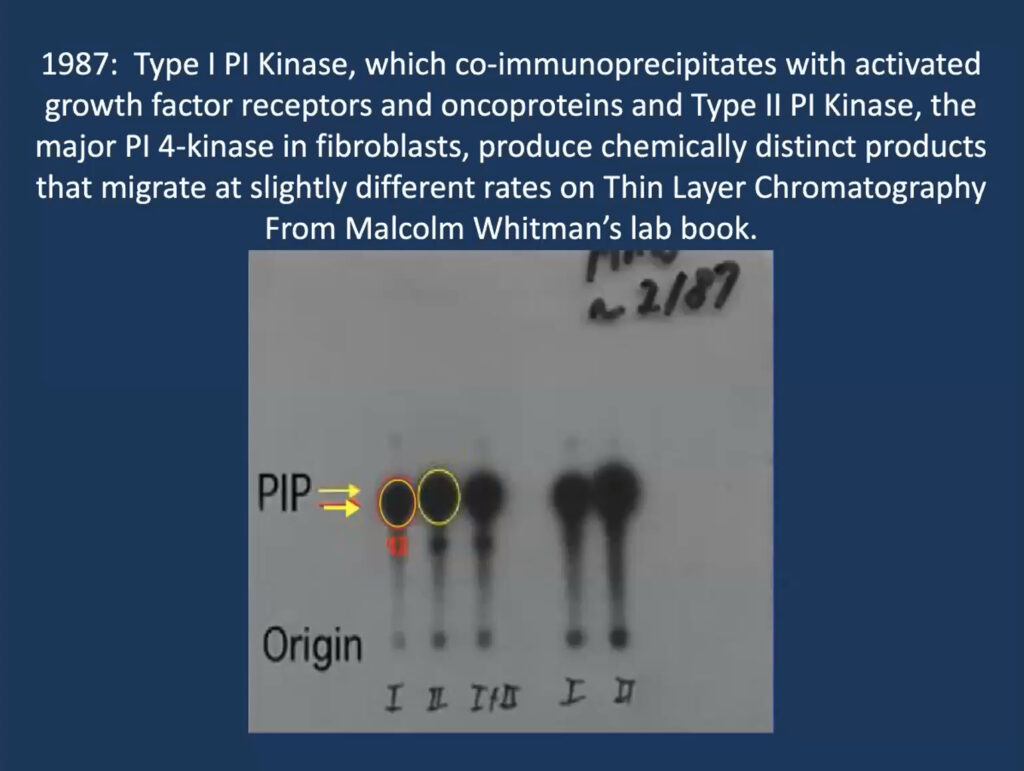
He traced his foundational work on PI3K inhibitors to a single slide that his then-graduate student, Malcolm Whitman, produced for a lab meeting in 1987 when he was at Tufts University in Boston. The lab was studying phosphatidylinositol and its role in a signaling cascade regulated to insulin and cell growth. They purified an insulin-activated lipid kinase and found that when activated, it is associated with proteins linked to cancer.
{kind=link}
At the time, researchers thought that phosphatidylinositol gets phosphorylated at position 4 on its sugar ring to produce a lipid called PI4P, which is involved in cell regulation. But Whitman found these phosphorylated lipids seemed to have two subtypes—one consistently ran a millimeter further on the gel. “That one-millimeter difference convinced me, as a chemist, that these had to be different species,” Cantley recalled. It turned out that their insulin-regulated kinase—now called PI3 kinase—produces a different molecule, called PI3P, and modulates a previously undiscovered insulin-regulated pathway that regulates the cell’s ability to take up glucose.
Cantley’s team went on to elucidate how PI3K regulates cells’ response to insulin signaling. “Almost every step in the first half of glycolysis is regulated by the PI3 kinase pathway,” he said. Collaborating with a team led by John Blenis, now at Weill Cornell Medicine, Cantley’s also fleshed out the connection between PI3K and multiple other cancer related signaling pathways, such as PTEN, Ras, and mTOR. Cantley called it “really quite remarkable” that “every tumor has at least one of the molecules in this pathway mutated—and many have several.” Indeed, accumulating research since then shows PI3K is one of the most frequently mutated oncogenes in cancer.
Although four different genes encode PI3Ks, just one of them—PIK3CA, which mediates insulin response, is widely mutated in cancers. Yet despite extensive effort, researchers have struggled to get PIK3CA-targeted drugs approved. Because this form of the protein mediates insulin activity, any drug that targets it makes patients insulin-resistant. “So you’re fighting the battle of insulin resistance as you are trying to treat the cancer, Cantley explained.
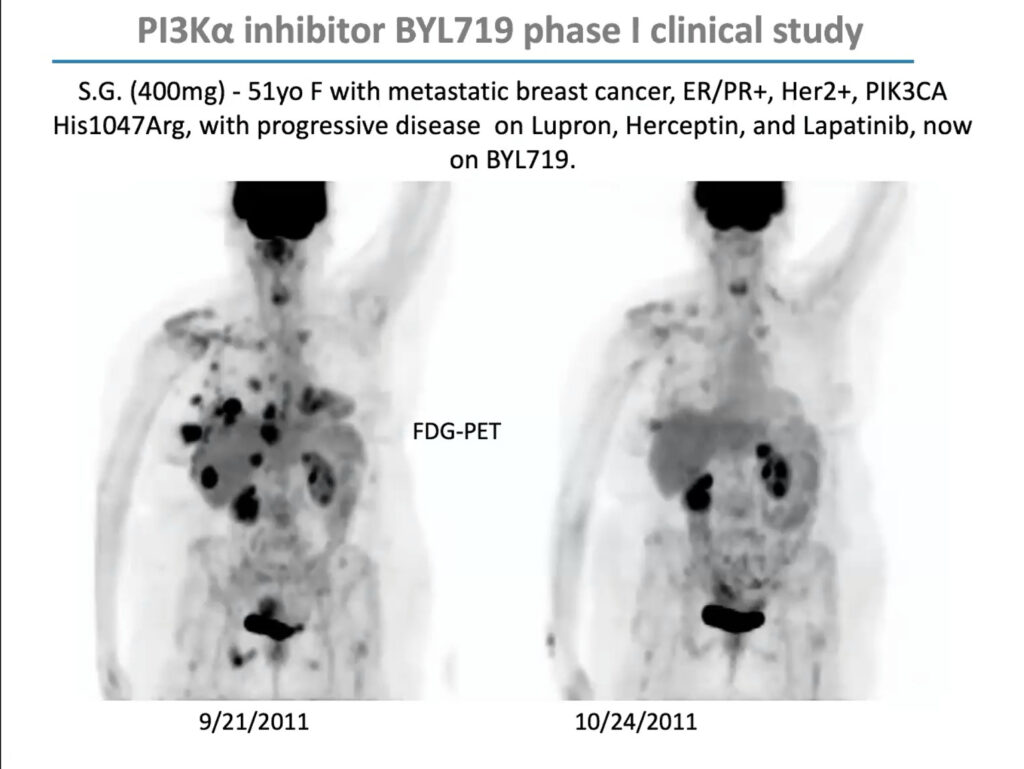
PIK3CA mutations are especially prevalent in uterine, cervical, breast, and ovarian cancer. In 2009, Cantley received a $12 million grant from the Stand Up To Cancer foundation to form a “dream team” of researchers to investigate how to develop PI3K inhibitors for women’s cancers and explore effective drug combinations. Through this grant, Cantley and his team conducted an early-stage trial of BYL719 (Alpelisib), a PIK3CA inhibitor developed by Novartis, in combination with a chemotherapy agent called letrozole, which lowers estrogen production, in people with metastatic breast cancer. The combination reduced glucose uptake to tumors and extended progression-free survival by roughly one year. The US Food and Drug Administration approved it in 2019.
{kind=link}
But in that early clinical trial, Cantley noticed that although the drugs’ response correlated with PIK3CA mutations overall, it was uneven; some patients with PIK3CA mutations didn’t respond, and some with mutations in unrelated genes did. “This was not a clear home run,” he said. “I argued, as we looked at the data, that maybe this has something to do with patients’ insulin levels.”
The insulin receptor (IR) is expressed in most tumors. Cantley and his colleagues wanted to interrupt the cycle in which high insulin levels trigger the IR and PI3K activity in the tumor, ultimately driving glucose uptake. To do so, the researchers tracked insulin levels in patients and mice taking PI3 kinase inhibitors. All three drugs they tested raised insulin levels 20-fold. In additional experiments, cells grown from patients with PIK3CA tumors died when exposed to PIK3CA inhibitors but were rescued when insulin was added. “So those ambient levels of insulin really are keeping the tumor alive,” Cantley said.
In subsequent studies, Cantley and his team tested the effects of insulin-lowering drugs, including metformin, as well as a ketogenic diet in mice. They found that a ketogenic diet effectively maintained low glucose and insulin levels while administering a PI3K inhibitor. In mice carrying tumors with a PIK3CA mutation, combining the PI3K inhibitor with a ketogenic diet significantly suppressed tumor growth compared to the control group. “This really tells us that keeping insulin down during the treatment with a PI3 kinase inhibitor could potentially have huge improvements in patient responses,” Cantley said.
Researchers are looking more closely at the effect of a ketogenic diet on the efficacy of PI3K inhibitors for endometrial cancer, breast cancer, and lymphoma. Previously a postdoc in Cantley’s lab and now at Weill Cornell Medicine, Marcus Goncalves is spearheading this effort with a trio of clinical trials currently enrolling patients.
Further Reading
Cantley
Whitman M, Downes CP, Keeler M, et al.
Nature. 1988; 323(6165):644-646.
Mayer IA, Abramson VG, Formisano L et al.
Clin Cancer Res. 2017; 23(1):26-34.
Goncalves MD, Hopkins BD, Cantley LC
Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer
N Engl J Med. 2018; 379(21):2052-2062.
Metabolic Pathways in Cancer
Speakers
Matthew Vander Heiden
Massachusetts Institute of Technology
Karen Vousden
Francis Crick Institute
Metabolic Limitations in Cancer
All cells in the body, including cancer cells, exist in different metabolic environments, and thus have different nutritional resources available to them, said Mathew Vander Heiden. Cancer cells have especially high metabolic needs because by definition, they proliferate—doubling the mass of proteins, nucleic acids, and lipids in order to go from one cell to two. Understanding how different types of cancer cells reorganize their metabolic pathways to accomplish this feat can bring insight into the role metabolism plays in cancer therapeutics.
Tissues solve their metabolic needs differently. Metabolism in brain cells, for example, differs from that in liver cells. These differences must be reflected in the gene expression patterns of the tissues’ metabolic networks. When a cell becomes cancerous, it takes its existing metabolic network, based on its environment, and reorganizes it to support its proliferation. That’s why cancers exhibit varied metabolisms and are sensitive to different therapies, Vander Heiden said.
His lab studies how a cell’s environment constrains its metabolic network. In one recent study, the researchers characterized the nutrients available in the interstitial fluid of pancreatic and lung tumors in mice. Transplanting a tumor into a different tissue site changed the nutrients available to it. But the mutational profile of a tumor had a much weaker effect on its metabolism. More recent human data suggests that nutrient profiles in the interstitial fluid of kidney cells and kidney tumor cells is similar. The findings suggest that nutrient availability is an intrinsic property of different tissues, and that cancer must adapt to the food available in a specific area. “That’s a cool idea, because it has a profound effect on how we think about metastasis,” said Vander Heiden. His lab has conducted metabolic phenotyping of mouse cancers transplanted to different tissue sites and found that the most significant outlier in the metabolic profile is in the brain. That matches clinicians’ experience with HER2-positive breast cancer, which tends to respond well to treatment except when the cancer reaches the brain.
To probe tissue-intrinsic metabolic responses, researchers in the lab of Rakesh Jain, also at Harvard, transplanted breast cancer tissue either into mammary fat pads or the brains of mice. They then treated the muse with PI3K inhibitor and found that the tumor shrunk in the mammary tissue but not the brain. Related studies showed that the brain microenvironment partially drove differences in response.
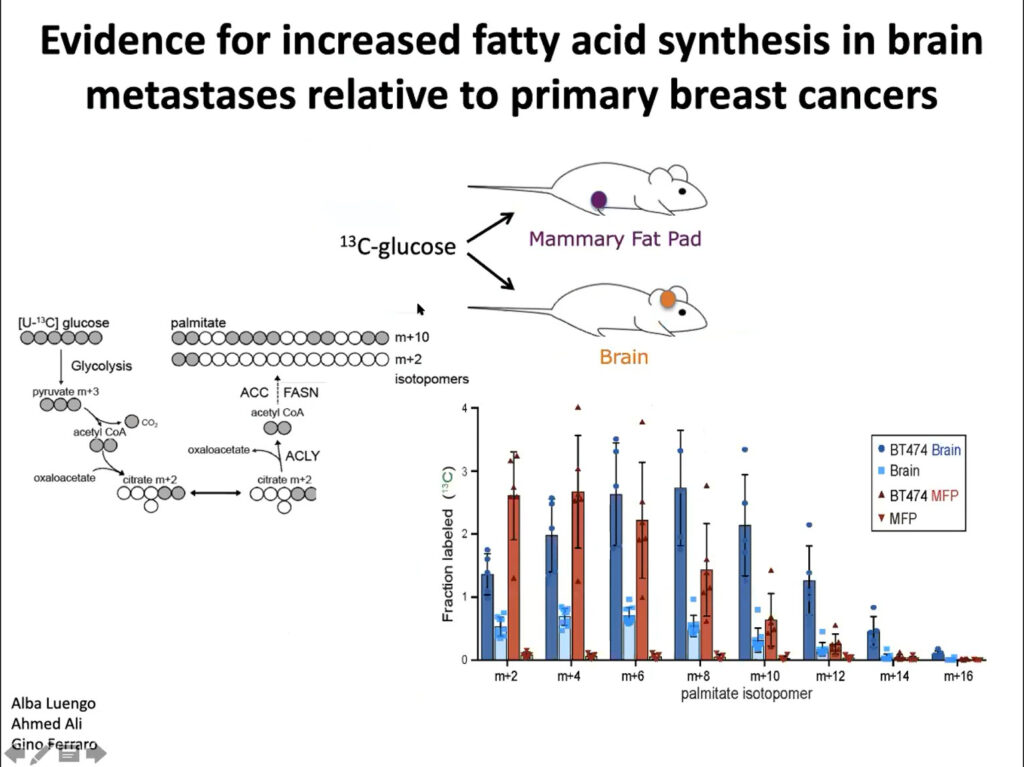
{kind=link}
In collaboration with Jain’s lab, Vander Heiden’s team implanted tumor cells into the brains and mammary fat pad tissue of mice. They found different metabolic gene expression and activity between the two sites. They also found more glucose uptake into fatty acids in the brain tumor tissue than in the mammary tissue, suggesting that fatty acid synthesis increases in brain metastases. Ultimately, they showed that these differences affect how tumors develop in the brain and facilitate tumor growth. The microenvironment and nutritional differences of tumor cells is key to understanding varied responses to metabolism-targeted therapies.
Metabolic Control of Tumor Progression
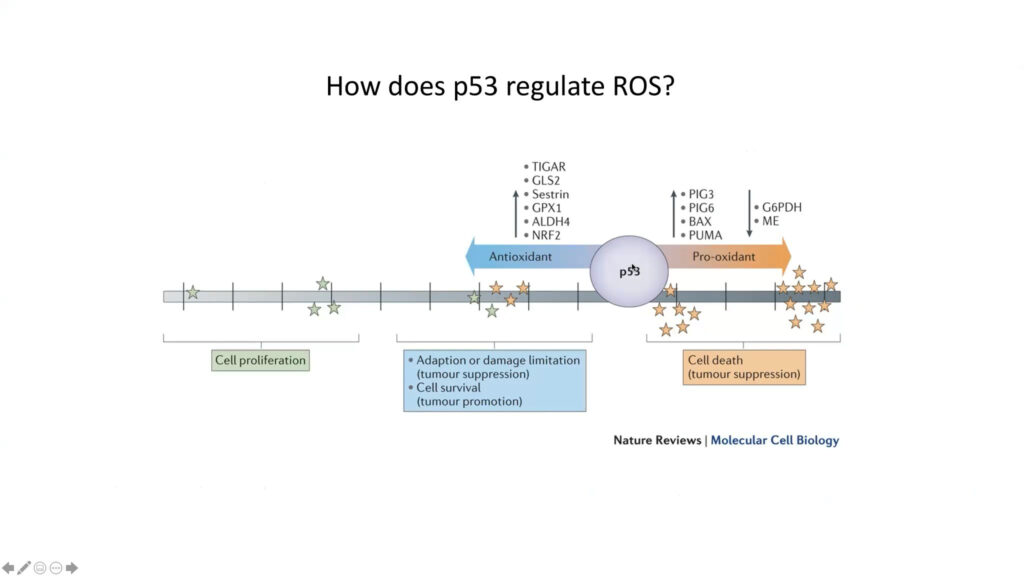
P53 is a key tumor suppressor gene. Some seven million cancer patients per year carry a mutant version of it. However, virtually all tumors, even those with a normal p53 gene, have lost p53’s tumor suppressive activity, said Karen Vousden. Early research showed that p53 suppresses tumors but activates cell death and senescence, killing tumor cells. But more recently, Vousden and others report another type of p53 activity, in which it contributes to the cell’s survival and its ability to adapt to stressful conditions, such as the accumulation of reactive oxygen species (ROS). Her lab is examining the gene’s role in metabolic signals such as nutrient fluctuation and oxidative stress.
Next, they looked at p53’s role in adapting to nutrient starvation. Comparing cell lines with p53 to without it, they found that p53 mitigates oxidative stress caused by cells lacking amino acids the body normally produces, such as serine. The absence of serine had stronger negative effects in cells without p53, reflecting the gene’s ability to drive antioxidant defense, Vousden said.
To look at how p53 protects cells from oxidation, the researchers focused on a downstream enzyme called TIGAR. Although the protein’s full suite of functions is still unknown, it promotes the production of NADPH, a molecule that supports antioxidant activity. Cells that lack TIGAR are more sensitive to oxidative stress and can’t maintain NADPH levels. Studies suggest that this effect is specific to mitochondria. Organoids lacking TIGAR have a much higher level of toxic ROS than organoids expressing the enzyme, and treating the cells with mitochondrial antioxidants — but not membrane antioxidant — mitigates this effect.
{kind=link}
Vousden’s lab also studied the effects of TIGAR during various stages of tumor development. They found that loss of TIGAR impedes tumor growth in both the pancreatic and intestinal adenoma models. Interestingly, mice with these tumors also had an increased rate of lung metastasis. Treating these mice with antioxidants mitigated the effect on lung metastasis, suggesting that ROS influences cancer development differently at different stages of the disease.
Further Readings
Vander Heiden
Leanne Li, Ng, SR, Colón CI, et al.
Identification of DHODH as a therapeutic target in small cell lung cancer
Sci Transl Med. 2019; 11(517):eaaw7852.
Dalin S, Sullivan MR, Lau AN.
Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance
Cancer Res. 2019; 79(22):5723-5733.
Halbrook CJ, Pontious C, Kovalenko I.
Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer
Cell Metab. 2019; 29(6):1390-1399.e6.
Vander Heiden MG, DeBerardinis RJ.
Understanding the Intersections between Metabolism and Cancer Biology
Cell. 2017; 168(4):657-669.
Vousden
Li H, G Jogl.
Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator)
J Biol Chem. 2009; 284(3):1748-54.
Cheung EC, DeNicola GM, Nixon.
Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer
Cancer Cell. 2020; 37(2):168-182.e4.
Humpton T, Vousden KH.
Taking up the reins of power: metabolic functions of p53
J Mol Cell Biol. 2019; 11(7):610-614.
The Clinical Outlook For Modulating Metabolism
Speaker
Ulrike Philippar
Johnson & Johnson
Targeting Metabolism and Differentiation Therapy in AML
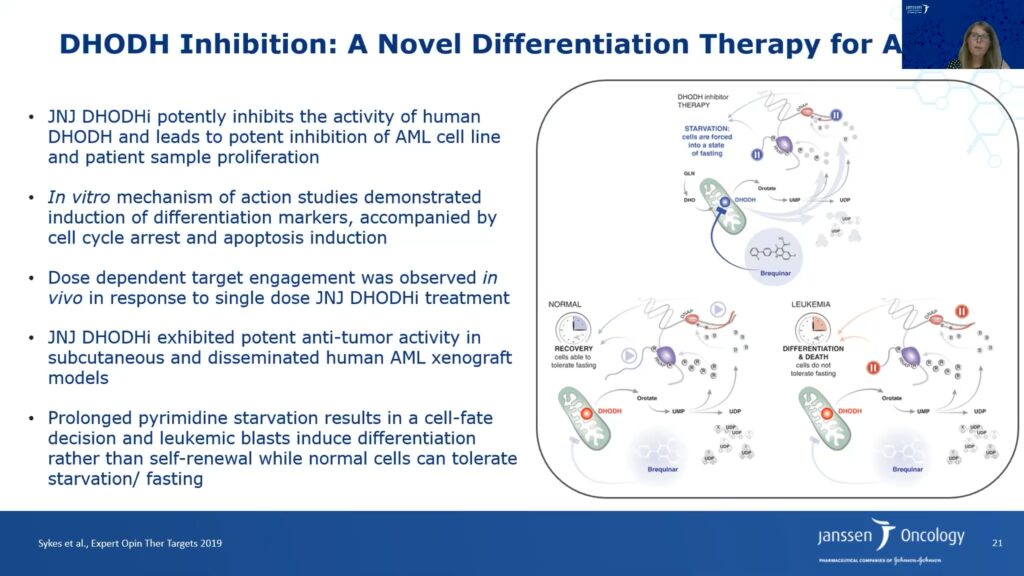
Acute Myeloid Leukemia (AML) is a critical disease area in Janssen’s drug discovery effort. A clinically heterogenous disease with multiple subtypes, AML is characterized by a diverse landscape of chromosomal abnormalities and gene mutations. One hallmark of AML, myeloid differentiation block, is linked to metabolism, said Ulrike Philippar. Myeloid differentiation in normal cells results in the generation of mature blood cells from hematopoietic stem cells. This process is blocked in AML cells, resulting in uncontrolled proliferation of progenitors, and ultimately, aggressive leukemia. Differentiation therapies have thus been a recent focus of drug development for AML, and unmet medical need for the disease remains high.
Philippar first described two US Food and Drug Administration-approved differentiation therapies. The first, called all-trans retinoic acid, or ATRA, was first used in 1985 to treat acute promyelocytic leukemia (APL), a subtype of AML. ATRA binds to a translocated protein in APL, leading to disruption of the differentiation block. The second therapy, IDH1 and IDH2 inhibitors, treats AML patients with an IDH mutation. The drug came to be used in combination with arsenic trioxide. These agents bind to a translocated protein in APL, disrupting the differentiation block.
Philippar and her team conducted two CRISPR/Cas9 screens to identify novel targets that inhibit proliferation in cell lines in vitro and/or in a bone marrow transplant mouse model in vivo. They identified dihydroorotate dehydrogenase (DHODH), an enzyme located in the mitochondrial membrane involved in pyrimidine synthesis. DHODH is also essential for the production of the nucleotide uridine monophosphate (UMP), the first building block in the production of RNA and DNA synthesis. Researchers, including Philippar and her colleagues, have shown that malignant cells, particularly AML cells, are metabolically dependent on pyrimidine production because they cannot salvage sufficient uridine from the extracellular environment.
Janssen identified a potent, selective proprietary DHODH inhibitor. The molecule strongly inhibited proliferation in 25 AML cancer cell lines. Janssen’s DHODH inhibitor also showed strong antiproliferative activity in primary AML samples. A few patient samples appeared to be resistant to the drug, and Janssen researchers will continue to investigate the basis of that resistance.
{kind=link}
The researchers also probed the DHODH inhibitor’s mechanism of action. The molecule induced cell cycle arrest in S phase, resulting in increased apoptosis. Furthermore, treatment with the DHODH inhibitor also increased cellular differentiation in AML cell lines. Adding excess uridine can reverse these effects, which validates the inhibitor’s on-target activity. Janssen’s DHODH inhibitor led to tumor regression in subcutaneous AML xenografts and increased the lifespan of mice bearing disseminated AML cancer cells approximately twofold.
Philippar said that DHODH inhibitor therapy takes away the pyrimidine source and leads to starvation of cells by forcing them into a fasting state. The researchers hypothesize that normal cells can tolerate fasting and recover; however, leukemia and other cancer cells cannot tolerate fasting because they require higher pyrimidine synthesis. Hence, prolonged pyrimidine starvation of leukemia cells results in a cell fate decision leading to differentiation, and ultimately, apoptosis and tumor regression.
DHODH confirms the relevance of metabolic targets for AML. However, the clinical diversity of AML may be a challenge for metabolic drugs, and researchers will have to better understand patient selection as they develop these drugs in the clinic.
Further Readings
Philippar
McKenzie MD, Ghisi M, Oxley EP, et al.
Interconversion between Tumorigenic and Differentiated States in Acute Myeloid Leukemia
Stem Cell. 2019;25(2):258-272.e9.
Stuani L, Sabatier M, Sarry J-E.
Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia
BMC Biol. 2019; 17(1):57.
Sykes DB, Kfoury YS, Mercier FE.
Cell 2016, 167(1):171-186.e15.
Sykes DB
Expert Opin Ther Targets. 2018; 22(11):893-898.
Panel Discussion
Panelists
Lewis C. Cantley, PhD
Weill Cornell Medical College
Matthew Vander Heiden, PhD
Massachusetts Institute of Technology
Karen Vousden, PhD
Francis Crick Institute
Ulrike Philippar, PhD
Johnson & Johnson
Panelists reflected on what moderator Costas Lyssiotis, a former postdoctoral student in Cantley’s laboratory, framed as a key theme of the symposium: harnessing diet as a way to treat cancer.
Cantley stated that previous efforts to use diet to understand cancer have largely failed, because getting trial participants to adhere to a diet and accurately report what they eat is nearly impossible. In a recent clinical trial that tested a ketogenic diet as a neoadjuvant for endometrial cancer, Cantley’s collaborator Marcus Goncalves used the metabolic kitchen at Weill Cornell Medical School to generate 21 meals per week for each trial participant. Weekly blood draws made it possible for researchers to be sure patients adhered to the diet. “To show [that] a dietary intervention works, you have to do it in a very controlled manner, not just tell people what they should eat and hope that they find it at the market,” Cantley said. Ultimately, the aim is to get dietary interventions approved as therapies so that insurance companies will cover them. He noted that companies such as Bayer and Novartis are excited by the possibility because it can potentiate pharmaceutical therapies. “We should hold diets to the same standards we hold drugs to,” said Cantley.
Vander Heiden agreed about the need for more rigorous dietary trials. His lab found that alterations in diet led to changes in the composition of metabolic molecules in interstitial fluid, and presumably those changes extend to nutrients available in different tissues and in blood. Animal studies are especially useful for identifying how nutrient availability changes with diet, he said; and they might point to unexpected differences beyond those already identified for the glucose and insulin pathway and serine.
Translating dietary interventions from animals to humans may not be straightforward. “What happens in mice doesn’t necessarily happen in people,” Vousden said. She and her colleagues are developing a serine-depleted diet for people with cancer, along the lines of low phenylalanine diets prescribed for children with phenylketonuria, an inherited condition that raises the level of the amino acid building block phenylalanine in the blood.
Any nutrient may be modulated for cancer therapy, but researchers must have a firm mechanistic understanding of why that alteration might work. “I think that’s at the heart of whether it will be successful, and whether people will follow these diets,” Vousden said. A solid grounding in the mechanism underlying a possible dietary intervention’s effect can help motivate patients to follow a restrictive diet for their clinical benefit.
Philippar noted that so far, considering diet is not standard practice in industry drug development. But it is potentially important, not just for targets that modulate the metabolism but also for immunological approaches. Researchers now know that diet can affect gut bacteria, which might in turn affect drug response. In clinical trials run by Janssen, she explained, researchers generally check whether a patient is in a fed or fasted state when receiving an experimental therapy, but they do not check on what exactly the patient has eaten.
Single-cell RNA sequencing makes it possible to look at the tumor microenvironment and to determine how changes in diet affect the expression levels of immune genes. In 2019, Cantley, Goncalves and others showed that just one, 12-ounce serving of a sugary drink such as orange juice, apple juice, or soda each day enhances intestinal tumors in mice. A combination of fructose and glucose causes the effect, the researchers found, and knocking out a molecule called ketohexakinase, which phosphorylates fructose, limits tumor growth. The connection to sugary drinks “is very surprising,” says Cantley, “but I think it may explain why we’ve almost tripped the rate of early onset colorectal cancer in young adults in the last 20 years.”