Neuroplasticity, Neuroregeneration, and Brain Repair
Reported by:
Alla Katsnelson
Presented by:
Eli Lilly and Company
The New York Academy of Sciences
Overview
Many promising strategies for promoting neuroregeneration have emerged in the past few years, but a further research push is needed for these ideas to be translated into therapies for neurodegenerative diseases.
On June 13–14, a symposium presented by Eli Lilly and Company and The New York Academy of Sciences brought together academic and industry researchers working on multiple neurodegenerative diseases as well as clinicians and government stakeholders to discuss cutting edge basic and clinical research on neuroregeneration and neurorestoration. Topics included neuronal plasticity, inflammation, glial cell function, autophagy, and mitochondrial function, as well as analysis of recent drug development failures and how to move forward from them.
Speakers
Benedikt Berninger, PhD,
University Medical Center Johannes Gutenberg University
Mainz, Germany
Graham Collingridge, PhD,
University of Toronto
Ana Maria Cuervo, MD, PhD,
Albert Einstein College of Medicine
Valina Dawson, PhD,
Johns Hopkins School of Medicine
Roman Giger, PhD,
University of Michigan
Steven Goldman, MD, PhD,
University of Rochester Medical Center
Eric Karran, PhD,
AbbVie
Arthur Konnerth, PhD,
Technical University of Munich, Germany
Guo-li Ming, MD, PhD,
Johns Hopskins School of Medicine
David Rowitch, MD, PhD, ScD,
University of Cambridge and University of California, San Fransisco
Amar Sahay, PhD,
Massachusetts General Hospital
Reisa A. Sperling, MD, MMSc,
Brighman and Women’s Hospital
James Surmeier, PhD,
Northwestern University
Richard Tsien, DPhil,
New York University, Longone Medical Center
Jeffrey Macklis,
Harvard University
Mark Mattson,
National Institute of Aging
Clive Svendsen,
Cedars-Sinai Medical Center
Michael Sofroniew,
David Geffen School of Medicine, UCLA
Michael J. O’Neill,
Eli Lilly and Company
Presented By


Meeting Reports
Meeting Reports
Astrocytes in CNS Repair; Disease-Modifying Therapies in the Pipeline
Speakers
Eric Karran
AbbVie
Highlights
- Astrocyte scar formation is not detrimental to neuronal regeneration and repair after injury but is in fact critical to the healing process.
- The clinical pipeline in Alzheimer’s disease is dominated by amyloid beta-targeting compounds, despite the fact that the approach has not been successful to date.
Astrocytes in CNS Repair
In his keynote talk, Michael V. Sofroniew of the University of California, Los Angeles, described 25 years of work on the overlooked and misunderstood role of astrocytes in the central nervous system (CNS).
These glial cells were discovered in the 19th century, and researchers widely believed that their activation after injury—which often results in scar formation around the lesion—detrimentally affects recovery. “But one has to ask, why would nature conserve this response to injury across all mammalian species if it were purely detrimental?” Sofroniew said.
Astrocytes can play fundamentally different roles in the CNS. In healthy tissue, they help synapses take up and release neurotransmitters and other factors, and help maintain neuronal energy balance and blood flow in surrounding tissue. Their activation in response to damage differs depending on whether recovery requires neurons to grow through lesioned tissue or through intact neural tissue.
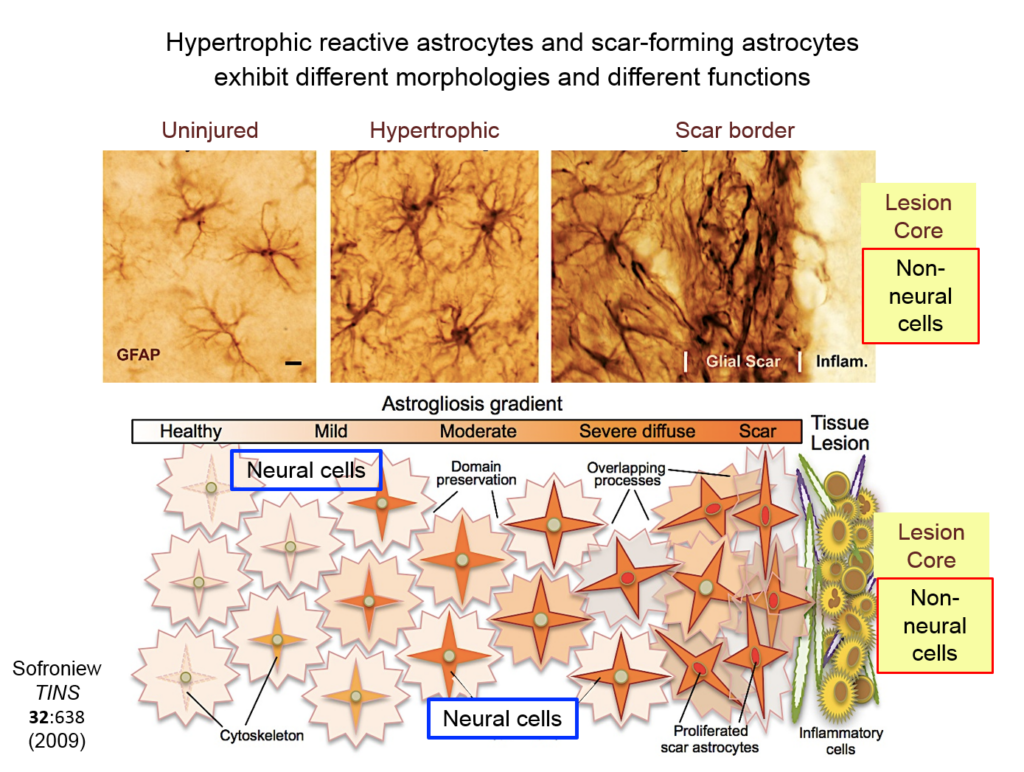
Astrocytes responding to injury exist in different phenotypes: a hypertrophic reactive form interacts with neural cells, and a scar-forming reactive form interacts with non-neuronal inflammatory and fibrotic cells. Researchers are just beginning to define the function of hypertrophic astrocytes, but Sofroniew and his colleagues hypothesize that they represent a beneficial gain of function—helping injured neurons make new synapses and reorganize damaged circuits. Much remains to be learned about this process, he said.
Ongoing research from Sofroniew’s lab suggests that scar-forming astrocytes have a different, also beneficial function: recruiting inflammatory cells into the tissue, regulating their activity, and restricting their spread outside the lesion. Inflammation is crucial for getting rid of damaged cells, but too much of it damages surrounding intact tissue.
When neural tissue is injured, astrocytes recruit cells to scavenge damaged tissue. Somehow, astrocytes sense where the border between damaged and healthy tissue should be and wall off the injury with scar tissue. Sofroniew and others have shown that disrupting scar formation causes neurons in surrounding tissue to die.
Entrenched dogma in the field, however, says that astrocyte scar formation prevents axon regeneration. Twenty years ago, Sofroniew’s lab first tested whether disrupting scar formation in mice would spur injured axons to spontaneously regenerate. Their results showed that it didn’t, but the findings went against current dogma so the team never published them. When a researcher interested in the question joined the lab recently, they began exploring the question again, using two mouse models with mutations that prevent scar format.
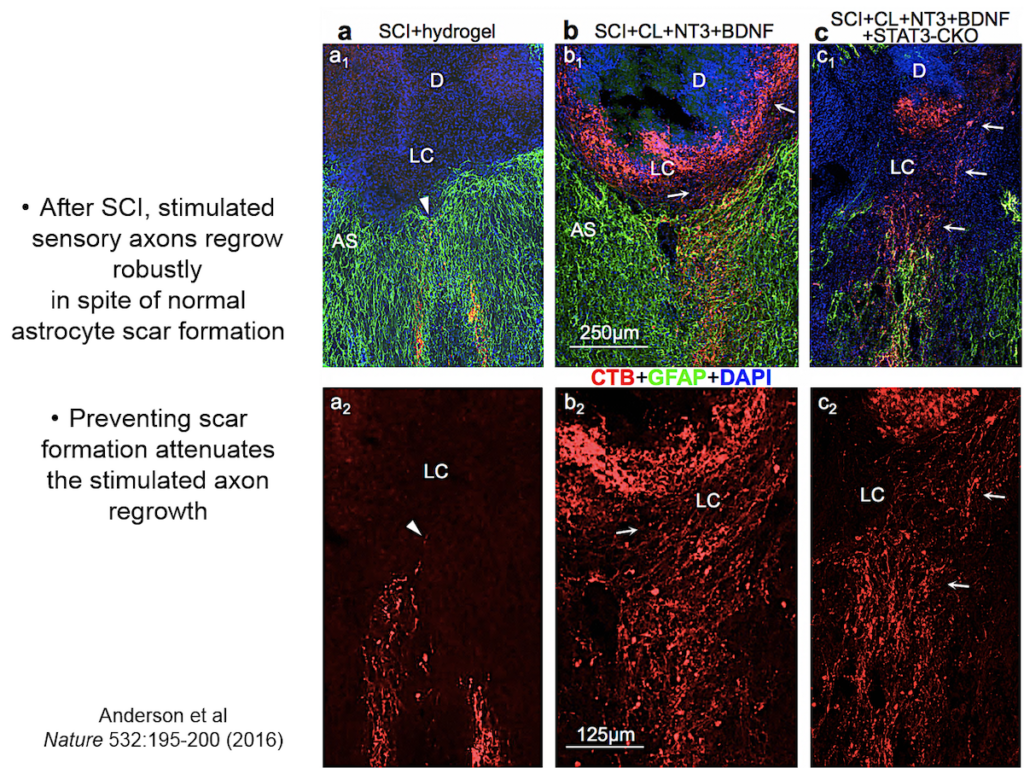
They showed that axons in three different types of CNS tracts failed to regrow in the mutant mice. Both astrocytes in lesions, along with other, non-astrocyte cells, all produced a variety of molecules both promoting and inhibiting axonal growth, underscoring the multi-component nature of regeneration. And axons that received appropriate stimulatory molecules “grow happily across astrocyte scars,” he said. The group is now confirming the result in additional types of CNS tracts. Sofroniew concluded that astrocyte reactivity and scar formation are not forms of astrocyte dysfunction, but adaptive functions critical for CNS repair and regeneration after injury.
Disease-Modifying Drugs for Alzheimer’s Disease: An Industry Perspective
The 1990s were a rich decade of discovery in Alzheimer’s disease, said Eric Karran of the pharmaceutical company AbbVie. Researchers identified disease-causing autosomal dominant mutations in the amyloid precursor protein presenilin and in tau. The field began to uncover key mechanisms and targets, and many believed that the next decade would yield effective therapeutics. However, that has not transpired, and many uncertainties about Alzheimer’s disease drug development remain.
Researchers still puzzle over the relationship between tau pathology and amyloid beta deposition. And while evidence suggests that Apolipoprotein E (ApoE) is closely involved in amyloid beta pathology, the mechanistic details remain mysterious. Nonetheless, research on the autosomal dominant mutations has geared drug discovery toward the idea that amyloid deposition initiates the disease process. Yet it is not clear that amyloid beta is an effective target for people who already have symptoms of Alzheimer’s disease.
Three questions are critical for therapeutics targeting amyloid: at what stage of the disease is such a drug most likely to be effective, by how much should amyloid beta be lowered, or its clearance be facilitated, and what kind of clinical experiment will test the validity of the amyloid cascade hypothesis.
Karran made a distinction between onset and duration of the disease. Possibly, amyloid beta deposition initiates the disease, he said, but is not the factor that drives its progression. The amyloid cascade hypothesis has many permutations, making proving or disproving it particularly difficult. One clear sign of this is the multiple failed trials that targeted amyloid beta. Lilly’s solanezumab seemed to show a mild effect on cognitive decline, but the signal was too small for a phase 3 trial. One currently promising candidate is Biogen’s aducanumab, which showed time- and dose-dependent reduction of amyloid plaques in early-stage trials.
A drug that intervenes with the onset and spread of tau pathology could potentially have therapeutic value relatively late in disease. Tau pathology is the most proximate marker for neuronal loss and cognitive impairment. Tau proteins are released by a currently unknown mechanism; how they are seeded and travel to distant neurons is also poorly understood. The process points to several points of interventions, such as anti-tau antibodies targeting seeds or fibrils. However, early efforts at tau therapeutics have failed.
Speaker Presentation
Further Readings
Michael Sofroniew
Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV.
Astrocyte scar formation aids central nervous system axon regeneration.
Nature. 2016 Apr 14;532(7598):195-200. doi: 10.1038/nature17623. Epub 2016 Mar 30.
Burda JE, Sofroniew MV.
Reactive gliosis and the multicellular response to CNS damage and disease.
Neuron. 2014 Jan 22;81(2):229-48. doi:10.1016/j.neuron.2013.12.034. Review.
Khakh BS, Sofroniew MV.
Diversity of astrocyte functions and phenotypes in neural circuits.
Nat Neurosci. 2015 Jul;18(7):942-52. doi: 10.1038/nn.4043.
Sofroniew MV.
Astrocyte barriers to neurotoxic inflammation.
Nat Rev Neurosci. 2015 May;16(5):249-63. doi: 10.1038/nrn3898. Review. Erratum in: Nat Rev Neurosci.
Eric Karran
Braak H, Del Tredici K.
Alzheimer’s disease: pathogenesis and prevention.
Alzheimers Dement. 2012 May;8(3):227-33. doi: 10.1016/j.jalz.2012.01.011. Epub 2012 Mar 30.
Braak H, Thal DR, Ghebremedhin E, Del Tredici K.
Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years.
J Neuropathol Exp Neurol. 2011 Nov;70(11):960-9. doi: 10.1097/NEN.0b013e318232a379.
Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, et al.
Lancet. 2016 Dec 10;388(10062):2873-2884. doi: 10.1016/S0140-6736(16)31275-2.
Karran E, De Strooper B.
The amyloid cascade hypothesis: are we poised for success or failure?
J Neurochem. 2016 Oct;139 Suppl 2:237-252. doi: 10.1111/jnc.13632.
Dendritic Spines, Axons, and Synapses in Neuroplasticity
Speakers
Richard Tsien
New York University Langone Medical Center
Roman J. Giger
University of Michigan School of Medicine
Jeffrey Macklis
Harvard University
Highlights
- Neuronal cell bodies regulate events at the synapse via the CamKII signaling pathway.
- Imperfect adaptation to the gradual loss of dopaminergic neurons in the striatum drives Parkinson’s disease symptoms
- Dectin1, a receptor expressed on the surface of macrophages, mediates a neuroregenerative immune response after injury.
- Growth cones may contain autonomous machinery for building the neuronal circuitry of the brain.
Regulation of Synapses and Synaptic Strength
Understanding the neural circuitry underlying learning and memory requires understanding how neurophysiological events at the synapse are integrated with molecular events in the nucleus such as gene transcription and protein translation, said Richard Tsien of New York University. At the synapse, this process depends on the combined activation of glutamate receptors and so-called L-type calcium channels. Tsien’s lab discovered that such dual activation is coordinated by the mobilization of a molecule called CamKII—known to be a key player in learning and memory—around tiny protrusions from dendrites called dendritic spines.
Tsien and his colleagues then elucidated how the signal from this synaptic activity is conveyed to the nucleus. Two of the four known forms of CamKII do their jobs at the synapse, but a third form, called gamma CamKII, shuttles calcium and its binding partner calmodulin to the nucleus, where it initiates a signaling cascade that drives the transcription of genes involved in long-term potentiation, a key molecular mechanism underlying learning and memory. Mice mutated to lack gamma-CamKII showed reduced learning and memory and did not upregulate key genes after training in memory tasks.
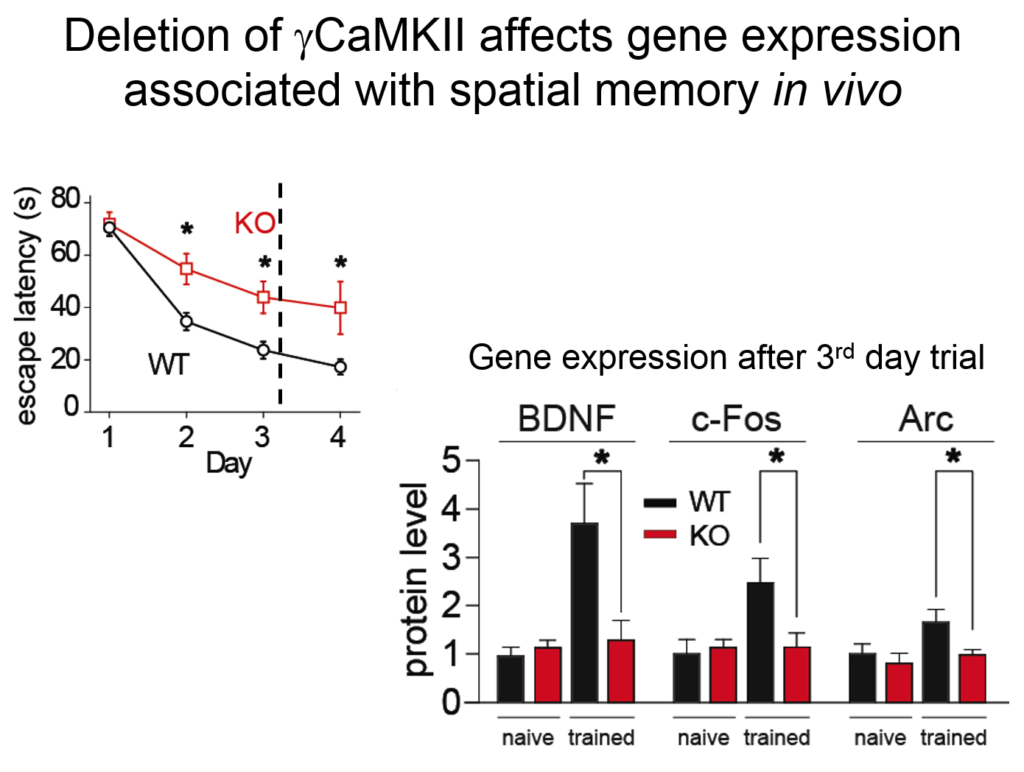
A mutation in gamma CamKII has been linked to intellectual disability in humans; studies on this human mutation revealed that it prevented the protein’s ability to shuttle calcium / calmodulin. Mutations in multiple proteins on this CamKII signaling pathway have been causally implicated in neuropsychiatric disorders such as autism, pointing to its importance in linking neuronal activity with nuclear processes.
Striatal Plasticity in Parkinson’s Disease
The core motor symptoms of Parkinson’s disease (PD) are caused by the loss of dopaminergic neurons in a brain region called the striatum. James Surmeier of Northwestern University described his lab’s research on how the two main pathways of the striatum—the direct (dSPN) and the indirect (iSPN) pathway—maintain homeostasis as the disease progresses.
Dopaminergic signaling in the striatum helps regulate goal-directed behaviors. The dSPN promotes desired actions, while the iSPN suppresses undesired actions, and the two must remain balanced for appropriate action selection to occur. Dopamine helps provide that balance. When its levels are high, it promotes long-term potentiation (LTP) of the dSPN (increasing choice of good actions) and long-term depression (LTD) of the iSPN (limiting opposition to them). When levels fall, the opposite occurs, quashing the selection of “bad” actions. Surmeier’s lab studies what drives LTP and LTD at these synapses by visualizing them. Only a subset of synapses is responsive to dopamine, they found.
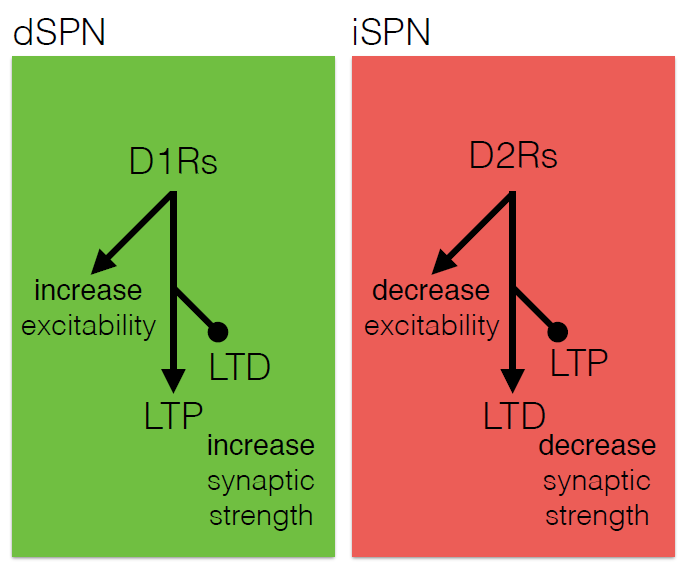
According to the standard model of Parkinson’s, loss of striatal neurons changes the excitability of the dSPN and iSPN, leading to suppression of motor activity. However, this model fails to account for how the system might compensate for its gradual deterioration. Such compensation may explain why the striatum must lose more than 60% of its dopaminergic cells before a person shows symptoms of the disease, Surmeier said. His work instead suggests that the dSPN and iSPN undergo a more graded but imperfect adaptation to the loss of dopaminergic innervation which distorts the information that these pathways receive, and which may cause deficits in goal-directed behavior before gross motor symptoms appear.
Immune-mediated Nervous System Regeneration
There is no spontaneous regeneration after nerve injury in the central nervous system. That is probably because extrinsic factors exist that block regeneration intrinsic factors that promote it are not activated, said Roman J. Giger of the University of Michigan School of Medicine. However, some types of inflammation can activate such regeneration factors.
His team found that an injection of zymosan (a mixture of proteins and carbohydrates prepared from the yeast cell wall) induced significant long-distance regeneration after optic nerve injury in mice, while the bacterial extract lipopolysaccharide did not. He and his colleagues found that this regenerative antifungal response is mediated primarily by a dectin-1, a receptor for a substance called beta glucan, which is expressed on the surface of macrophages and other immune cells, as well as by the immune recognition protein Toll-like receptor 2 (TLR2).
They also found this mechanism in spinal cord regeneration, as tested after a so-called conditioning injury to the sciatic nerve (which activates immune response genes) followed by a spinal cord lesion at the dorsal root ganglion. Wild type mice showed significant spinal cord axon regrowth after zymosan injection, while mice engineered to lack dectin-1 or TLR2 showed none.

The researchers then tried to pinpoint which immune cell types produced dectin-1, and where it had to be localized to spur regeneration. They found that immune cells from the sciatic nerve—that is, the conditioning lesion—carried the signal. Although mice lacking dectin showed no regeneration, immune cells from the lesioned sciatic nerve of a wild type mouse transplanted into the dectin-1 knockout mouse could rescue this deficit.
Growth Cone Control over Circuit Development
Building the brain’s neuronal circuitry is enormously complex endeavor: neurons exist in a multitude of diverse subtypes, they project to precise sompatotopic targets, and some send projections to more than one specific location. Projections can be up to a meter in length – some 10,000 cell body diameters away. The system’s precision is astounding, said Jeffrey Macklis of Harvard University, and being able to rebuild circuits when they go awry is key to regeneration in the face of injury or disease.
Macklis described work showing that the transcriptional machinery that generates this complexity is present not just in the neuronal cell body, but also in growth cones located at the tips of projections as they extend. His lab has found that growth cones contain locally translated proteins, suggesting that these neuronal outposts might exert autonomous control over circuit development. “As a developmentalist, I view growth cones as little baby synapses,” Macklis said.
Immature axons transplanted in the developing mouse still project to their original, appropriate targets, suggesting a logic and subtype specificity to the process. Macklis’s lab came up with an approach to label and isolate growth cones from different neuronal subtypes. They found specific protein and RNA enriched at growth cones that was not present in the neuronal cell body, suggesting a localized projection machinery. Targeting this machinery could be an important strategy for promoting regeneration.
Speaker Presentations
Further Readings
Richard Tsien
Barco A, Alarcon JM, Kandel ER.
Cell. 2002 Mar 8;108(5):689-703.
Deisseroth K, Heist EK, Tsien RW.
Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons.
Nature. 1998 Mar 12;392(6672):198-202.
Li B, Tadross MR, Tsien RW.
Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression.
Science. 2016 Feb 19;351(6275):863-7. doi: 10.1126/science.aad3647.
Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, Tsien RW.
γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression.
Cell. 2014 Oct 9;159(2):281-94. doi: 10.1016/j.cell.2014.09.019.
James Surmeier
Gerfen CR, Surmeier DJ.
Modulation of striatal projection systems by dopamine.
Annu Rev Neurosci. 2011;34:441-66. doi: 10.1146/annurev-neuro-061010-113641. Review.
Surmeier DJ, Graves SM, Shen W.
Dopaminergic modulation of striatal networks in health and Parkinson’s disease.
Curr Opin Neurobiol. 2014 Dec;29:109-17. doi: 10.1016/j.conb.2014.07.008. Epub 2014 Jul 22.
Surmeier DJ, Obeso JA, Halliday GM.
Selective neuronal vulnerability in Parkinson disease.
Nat Rev Neurosci. 2017 Jan 20;18(2):101-113. doi:10.1038/nrn.2016.178. Review.
Thiele SL, Chen B, Lo C, Gertler TS, Warre R, Surmeier JD, Brotchie JM, Nash JE.
Neurobiol Dis. 2014 Nov;71:334-44. doi: 10.1016/j.nbd.2014.08.006. Epub 2014 Aug 27.
Roman Giger
Baldwin KT, Carbajal KS, Segal BM, Giger RJ.
Neuroinflammation triggered by β-glucan/dectin-1 signaling enables CNS axon regeneration.
Proc Natl Acad Sci USA. 2015 Feb 24;112(8):2581-6. doi: 10.1073/pnas.1423221112. Epub 2015 Feb 9.
Jeffrey Macklis
Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD.
Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo.
Neuron. 2005 Jan 20;45(2):207-21.
Cederquist GY, Azim E, Shnider SJ, Padmanabhan H, Macklis JD.
Lmo4 establishes rostral motor cortex projection neuron subtype diversity.
J Neurosci. 2013 Apr 10;33(15):6321-32. doi: 10.1523/JNEUROSCI.5140-12.2013.
Inflammation, Oxidative Stress, Mitochondrial Function, and Autophagy
Speakers
Highlights
- Fasting and exercise exert protective effects on the brain and improve the bioenergetics properties of neurons.
- Activators of a selective autophagy process may help clear aggregating proteins implicated in neurodegenerative disease.
- A key cluster of Parkinson’s disease proteins regulate mitochondrial biogenesis and function.
Bioenergetic Challenges Bolster Brain Resilience
Mark P. Mattson of the National Institute of Aging described how two bioenergetics challenges—food deprivation and exercise—affect brain health. The ability to function under conditions of food deprivation is the main driving force in brain evolution, he said: Fasting was frequent, and it drove humans to search for food. Aging is a major risk factor for dementia and stroke, but sedentary lifestyles contribute as well, by compromising cells’ ability to adapt to the molecular stresses of aging.
Increased exercise is known to boost brain levels of the neuroprotective factor BDNF, and early work in Mattson’s lab found that fasting has the same effect in mice. Also, in mice genetically engineered to be obese and diabetic, alternate day fasting and increased exercise on a running wheel increased the density of synaptic spines in their brain. Further work showed that fasting and exercise also increased the number of mitochondria—the cell’s energy-generating organelles—in cultured hippocampal neurons.
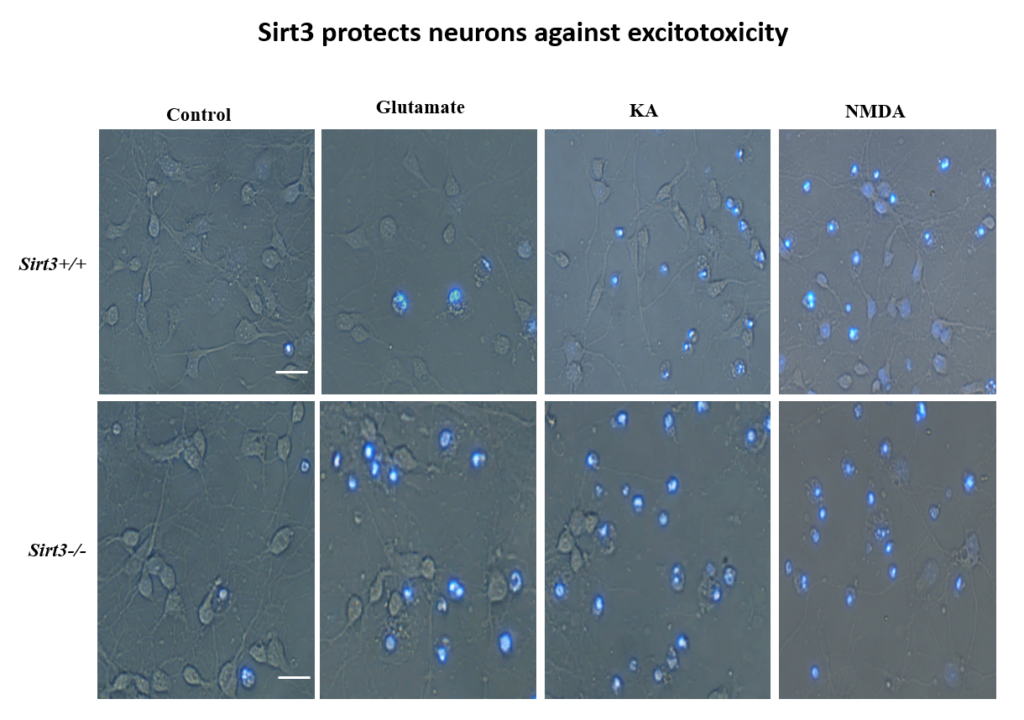
More recently, Mattson’s lab found that exercise and intermittent fasting upregulate a mitochondrial protein called sirtuin 3 (sirt3), which goes on to block enzymes that protect the mitochondria against stress and protect cells against apoptosis. The group has also explored the effects of fasting in humans. Currently, the group is studying whether people at risk for cognitive impairment due to age or metabolic status benefit from fasting two days per week.
Malfunctioning Autophagy Pathways in Neurodegeneration
Autophagy is the process of degradation or recycling of materials inside the cell, and many facets of it are coming under scrutiny as causal factors in neurodegeneration. Ana Maria Cuervo of the Albert Einstein College of Medicine studies chaperone-mediated autophagy (CMA), in which individual proteins targeted with a degradation motif are recognized by a chaperone protein, carried to a receptor called LAMP-2A on the lysosome surface, and pulled inside for degradation. In order to study the role of CAM in neurodegeneration, Cuervo’s lab designed a fluorescent reporter system that can track the process in vivo, in the brain and other organs.
The CAM pathway is highly sensitive to aging; levels of the LAMP-2A receptor drop as animals age. Additionally, many proteins involved in neurodegenerative diseases have CMA degradation motifs. The mutant form of LRRK2, the protein most often mutated in familial cases of Parkinson’s, interferes with LAMP-2 receptor’s ability to form complexes as required for translocation into the lysosome; other neurodegeneration-related proteins, such as tau, showed a similar effect, which led to an aggregation of these proteins due to their inability to be broken down inside the lysosome. Human postmortem Alzheimer’s disease brains also appear to have a CMA deficit.
The lab has now developed a selective activator of the CAM pathway and is administering it to a mouse model of Alzheimer’s disease. The intervention ameliorates behavioral symptoms such as anxiety, depression, and visual memory in the animals, as well as cellular markers of the disease.
Mitochnodrial Mechanisms and Therapeutic Opportunities
Mitochondrial dysfunction was first observed in Parkinson’s disease some 40 years ago, but how it plays a role in the disease is unknown. Some genetic causes of PD have been identified, including mutations in Parkin and PINK1. Valina L. Dawson’s lab at Johns Hopkins University is investigating how three closely interacting proteins, Parkin, PINK1, and PARIS, regulate mitochondrial function and, in turn, the integrity of dopaminergic neurons, which malfunction in PD.
In 2011, Dawson’s lab identified PARIS, a protein that tamps down mitochondrial production by repressing another protein called PGC1-alpha. PARIS is ubiquitinated by Parkin to remove the brake on mitochondrial production. Mice genetically engineered to lack Parkin show age-dependent loss of dopaminergic neurons and serve as a model of PD. But if these mice also experience a knock-down in PARIS, the deficit is rescued. Loss and gain of function studies of these proteins in mice revealed a homeostasis between them that regulates mitochondrial biogenesis and function. Pink1 is also central; it must phosphorylate Parkin for this homeostasis to occur.
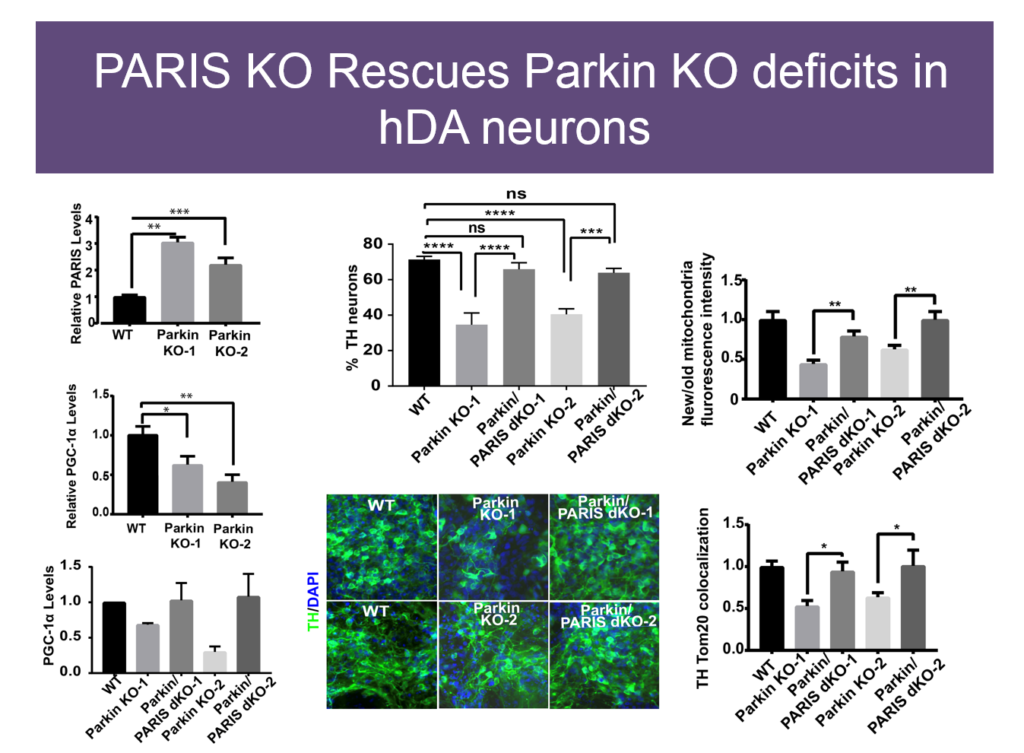
The relationships between these proteins also hold in human embryonic stem cells when these proteins are knocked down, and in induced pluripotent cells derived from Parkinson’s patients with mutations in these proteins. Based on these findings, Dawson’s team and collaborators are exploring whether PARIS inhibitors, Parkin activators, or other molecules affecting this protein network have therapeutic value in PD mice.
Speaker Presentations
Further Readings
Mark Mattson
Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, Bohr VA, Mattson MP.
Cell Metab. 2016 Jan 12;23(1):128-42. doi: 10.1016/j.cmet.2015.10.013. Epub 2015 Nov 19.
Mattson MP.
Energy intake and exercise as determinants of brain health and vulnerability to injury and disease.
Cell Metab. 2012 Dec 5;16(6):706-22. doi: 10.1016/j.cmet.2012.08.012. Epub 2012 Nov 15. Review.
Mattson MP, Longo VD, Harvie M.
Impact of intermittent fasting on health and disease processes.
Ageing Res Rev. 2016 Oct 31. pii: S1568-1637(16)30251-3. doi: 10.1016/j.arr.2016.10.005. [Epub ahead of print] Review.
Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP.
Nat Neurosci. 2008 Mar;11(3):309-17. doi: 10.1038/nn2055. Epub 2008 Feb 17.
Ana Maria Cuervo
Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM.
Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives.
Nat Chem Biol. 2013 Jun;9(6):374-82. doi: 10.1038/nchembio.1230. Epub 2013 Apr 14.
Wong E, Cuervo AM.
Autophagy gone awry in neurodegenerative diseases.
Nat Neurosci. 2010 Jul;13(7):805-11.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D.
Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy.
Science. 2004 Aug 27;305(5688):1292-5.
Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM.
Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease.
J Neurosci. 2011 Dec 14;31(50):18492-505. doi: 10.1523/JNEUROSCI.3219-11.2011.
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E,
Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM.
Interplay of LRRK2 with chaperone-mediated autophagy.
Nat Neurosci. 2013 Apr;16(4):394-406. doi: 10.1038/nn.3350. Epub 2013 Mar 3.
Valina Dawson
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM.
PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease.
Cell. 2011 Mar 4;144(5):689-702. doi: 10.1016/j.cell.2011.02.010.
Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, Shin JH, Dawson TM.
Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration.
Proc Natl Acad Sci USA. 2015 Sep 15;112(37):11696-701. doi: 10.1073/pnas.1500624112. Epub 2015 Aug 31.
Lee Y, Stevens DA, Kang SU, Jiang H, Lee YI, Ko HS, Scarffe LA, et al.
PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival.
Cell Rep. 2017 Jan 24;18(4):918-932. doi: 10.1016/j.celrep.2016.12.090.
Glial Function
Speakers
Highlights
- Glial cell dysfunction may causally contribute to schizophrenia and other neurological diseases.
- Astrocytes engineered to produce GDNF are in clinical trials for treating amyotrophic lateral sclerosis.
- Astrocytes are functionally and regionally heterogeneous, and their dysfunction may contribute to neurodegenerative disease.
Targeting Glial Cell Dysfunction in Neurological Disease
Glial cells make up a significant proportion of cells in the brain, yet their contribution to disease is poorly understood. Steven A. Goldman of the University of Rochester Medical Center studies glia’s role in brain diseases such as schizophrenia. His lab injects human glial progenitor cells into the brains of mutant mice that lack their own glia; the brains of the resultant chimeras become fully repopulated with human astrocytes and oligodendrocytes. This human glial chimera maintains the phenotypes of human glial cells, and mice with human glia show stronger long-term potentiation in the hippocampus and learn fear-conditioning and other behavioral and cognitive tasks more quickly than wildtype mice.
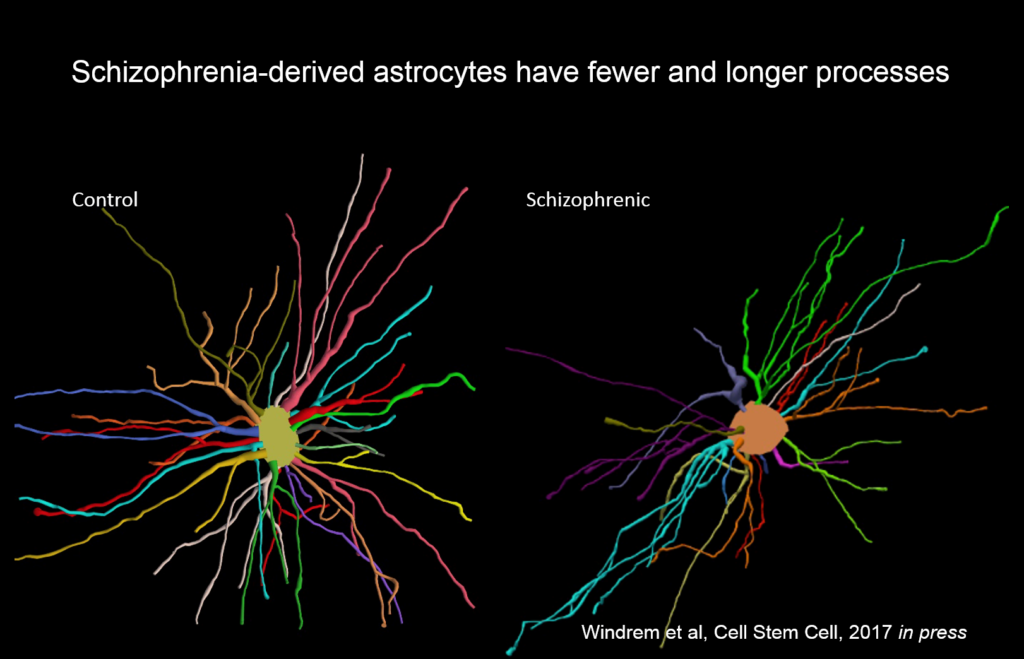
Goldman’s team created chimeric mice populated by glia derived from eight different people with juvenile onset schizophrenia, and compared them to mice with glial cells derived from control subjects. These glial precursor cells migrated abnormally and formed less myelin than precursors from control human subjects. Myelin-producing and glial differentiation genes, as well as genes associated with synaptic development and transmission, were downregulated. Astrocytes in the patient-derived chimeras also had irregular morphology. The animals exhibited impaired response to stimuli as well as anxiety and antisocial behavior. Genes related to glial cells might be potent therapeutic targets for schizophrenia and other diseases, like Huntington’s disease and frontotemporal dementia.
“We never thought of these as glial diseases, but fundamentally they might be,” Goldman said.
Stem-cell-derived Astrocytes for Treating Neurodegenerative Disease
Ninety percent of neurodegenerative diseases have no known genetic cause, and may be amenable to treatment with cell therapy, said Clive Svendsen of Cedars-Sinai Medical Center. While delivering neurons into diseased CNS is still evolving, astrocytes have great potential for immediate use, Svendsen said.
His lab developed a protocol for deriving astrocytes from human fetal tissue; these cells migrate to areas of damage when delivered to a rat brain. To give these cells more regenerative capacity, Svendsen and collaborators engineered the cells to release the growth factor GDNF. They initially tested this cell delivery therapy in a Parkinson’s disease model, but it has also been applied in stroke, and both Huntington’s and Alzheimer’s disease.
More recently they have begun to explore its use in amyotrophic lateral sclerosis (ALS), where life expectancy after diagnosis is a mere three years and no treatments exist. They first tested it in an ALS rat transgenic model in which astrocytes lacked the protein SOD1. When they transplanted the therapeutic astrocytes to the lumbar spine, the cells survived well and improved neuronal survival, but did not prevent paralysis. As they moved up the spinal cord, results improved; cell delivery into the brain’s motor cortex yielded improved motor function and survival in the animals.
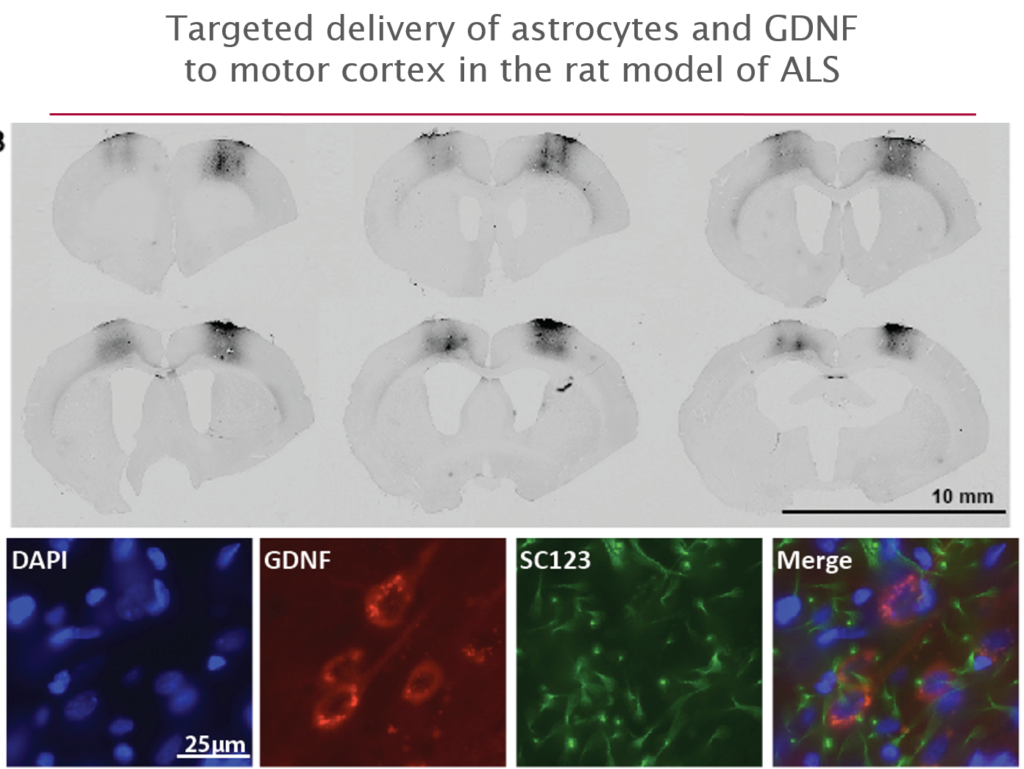
Last October, Svendsen and his team launched an 18-person clinical trial of this approach. For safety reasons, the U.S. Food and Drug Administration required the researchers to start by delivering cells into the lumbar spine; patients will receive the therapy in one leg, with the other acting as a control. If the first few patients experience no adverse effects, delivery into the cervical spine and the cortex will also be attempted.
Functionally Heterogeneous Astrocytes in the Mammalian CNS
How neuron patterning generates a diversity of neuronal types throughout the central nervous system is well understood. But very little is known about heterogeneity in astrocytes, although they are the most abundant cells in the CNS, comprising about half of all brain cells, said David H. Rowitch of the University of Cambridge.
Early work in Rowitch’s lab identified an astrocyte-specific transcription factor that showed that astrocytes are allocated to specific regions of the brain during development. They then searched for postnatal astrocytes in the spinal cord that were regionally and functionally distinct by comparing gene expression in the dorsal and ventral part of the spinal cord. The gene Sema3a was most highly expressed in ventral astrocytes in mice, and when it was deleted, half the animal’s alpha motor neurons, which innervate fast-twitching muscle, died.
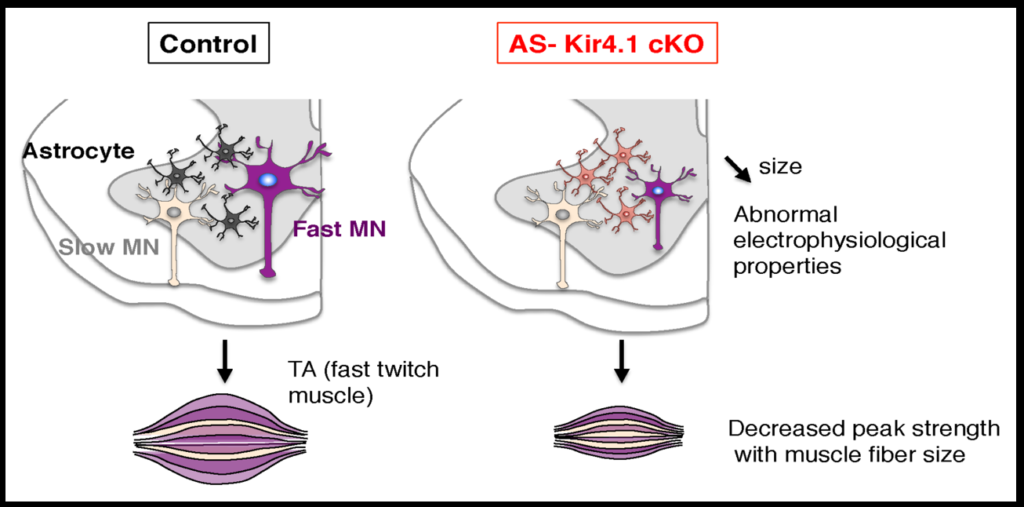
To investigate how neurons and astrocytes interact, the researchers examined a potassium channel called Kir4.1, which is preferentially expressed in the ventral brain and spinal cord. Loss of function mutations to the channel cause epilepsy, and the channel is strongly downregulated in astrocytes of people with ALS. Mice engineered to lack the channel in astrocytes have smaller alpha motor neurons and weaker muscle function. Transfecting the astrocytes of these mice with the channel reverses these deficits. The fact that astrocytes so strongly affect neuron function suggests that dysfunction in specific subsets of astrocyte may play a role in neurodegenerative diseases.
Speaker Presentations
Further Readings
Steven Goldman
Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L,
Betstadt S, Silva AJ, Takano T, Goldman SA, Nedergaard M.
Cell Stem Cell. 2013 Mar 7;12(3):342-53. doi: 10.1016/j.stem.2012.12.015.
Windrem MS, Schanz SJ, Guo M, Tian GF, Washco V, Stanwood N, Rasband M, Roy NS, Nedergaard M, Havton LA, Wang S, Goldman SA.
Cell Stem Cell. 2008 Jun 5;2(6):553-65. doi: 10.1016/j.stem.2008.03.020.
Osorio MJ, Goldman SA.
Glial progenitor cell-based treatment of the childhood leukodystrophies.
Exp Neurol. 2016 Sep;283(Pt B):476-88. doi: 10.1016/j.expneurol.2016.05.010. Epub 2016 May 8.
Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, Goldman SA.
Cell Stem Cell. 2013 Feb 7;12(2):252-64. doi: 10.1016/j.stem.2012.12.002.
Windrem MS, Schanz SJ, Morrow C, Munir J, Chandler-Militello D, Wang S, Goldman SA.
J Neurosci. 2014 Nov 26;34(48):16153-61. doi: 10.1523/JNEUROSCI.1510-14.2014.
Clive Svendsen
Capowski EE, Schneider BL, Ebert AD, Seehus CR, Szulc J, Zufferey R, Aebischer P, Svendsen CN.
J Neurosci Methods. 2007 Jul 30;163(2):338-49. Epub 2007 Mar 3.
Ebert AD, Barber AE, Heins BM, Svendsen CN.
Exp Neurol. 2010 Jul;224(1):155-62.
Ebert AD, Beres AJ, Barber AE, Svendsen CN.
Gowing G, Shelley B, Staggenborg K, Hurley A, Avalos P, Victoroff J, Latter J,
Garcia L, Svendsen CN.
Neuroreport. 2014 Apr 16;25(6):367-72. doi: 10.1097/WNR.0000000000000092.
Suzuki M, Svendsen CN.
Combining growth factor and stem cell therapy for amyotrophic lateral sclerosis.
Trends Neurosci. 2008 Apr;31(4):192-8. doi: 10.1016/j.tins.2008.01.006. Epub 2008 Mar 10.
Svendsen CN, Clarke DJ, Rosser AE, Dunnett SB.
Exp Neurol. 1996 Feb;137(2):376-88.
Thomsen GM, Gowing G, Latter J, Chen M, Vit JP, Staggenborg K, Avalos P, Alkaslasi M, Ferraiuolo L, Likhite S, Kaspar BK, Svendsen CN.
J Neurosci. 2014 Nov 19;34(47):15587-600. doi: 10.1523/JNEUROSCI.2037-14.2014.
David Rowitch
Freeman MR, Rowitch DH.
Evolving concepts of gliogenesis: a look way back and ahead to the next 25 years.
Neuron. 2013 Oct 30;80(3):613-23. doi: 10.1016/j.neuron.2013.10.034. Review.
Molofsky AV, Kelley KW, Tsai HH, Redmond SA, Chang SM, Madireddy L, Chan JR, Baranzini SE, Ullian EM, Rowitch DH.
Astrocyte-encoded positional cues maintain sensorimotor circuit integrity.
Nature. 2014 May 8;509(7499):189-94. doi: 10.1038/nature13161.
Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, Tenney A, Murnen AT, Fancy SP, Merkle F, Kessaris N, Alvarez-Buylla A, Richardson WD, Rowitch DH.
Regional astrocyte allocation regulates CNS synaptogenesis and repair.
Science. 2012 Jul 20;337(6092):358-62. doi: 10.1126/science.1222381. Epub 2012 Jun 28.
Innovative Approaches to Promote Neuroregeneration
Speakers
Highlights
- Novel therapies targeting the synaptic plasticity pathways could address the dysregulation of long term depression underlying Alzheimer’s disease.
- Brain organoids grown from human induced pluripotent stem cells recapitulate development and can model brain disease.
- Reprogramming pericyte cells into neuronal cells occurs via a distinct developmental program.
- Promoting neurogenesis and re-engineering molecular connectivity in the hippocampus restored age-related memory decline in mice.
Is Alzheimer’s Disease Caused by Long Term Depression Gone Awry?
One key purpose of brains is to enable learning and memory—a process that relies on a balance between long term potentiation (LTP) and long term depression (LTD) to drive synaptic plasticity, said Graham Collingridge of the University of Toronto. Dysregulation of that balance causes Alzheimer’s disease, he said.
In 1983, Collingridge’s lab identified the role of the NMDA receptor in synaptic plasticity, finding that its activation could cause both LTP and LTD. In later work, they sought kinase inhibitors that could block LTP and LTD. One of the few ways to inhibit LTD was to block glycogen synthase kinase 3beta (GSK-3beta). This molecule is also known as tau kinase because it hyperphosphorylates the protein tau—a process implicated in Alzheimer’s disease pathogenesis. “I thought, well, that’s just not coincidence, is it,” Collingridge said.
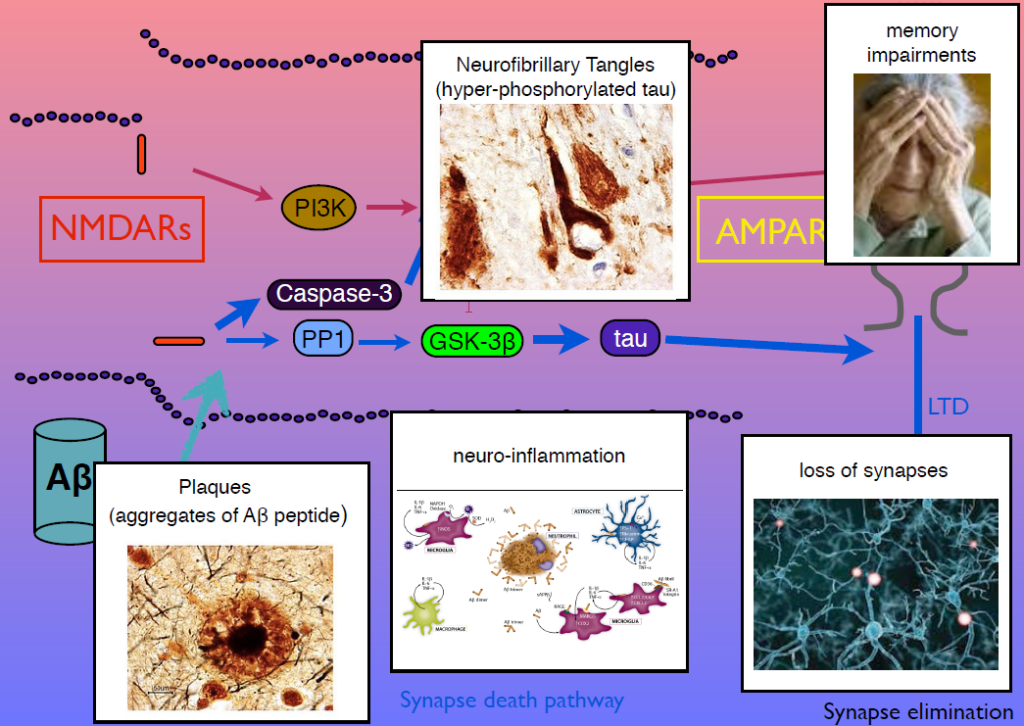
Tau regulates microtubules in axons, but Collingridge’s lab found that it also exists in synapses, and is phosphorylated by GSK-3beta. In mice engineered to lack tau, LTD is absent but LTP is undisturbed. Work from other researchers had shown that amyloid beta, the protein that aggregates in Alzheimer’s disease, inhibits LTP and facilitates LTP. His group showed that GSK-3beta reverses this effect, and identified other parts of the signaling pathway linking amyloid beta, tau, GSK-3beta, and both LTP and LTD. Dysregulation in these components can generate amyloid beta plaques, tau tangles, and the neuroinflammation, synapse loss and memory loss that characterizes Alzheimer’s. Modulators of NMDA receptor activity may have therapeutic potential.
Modeling Human Brain Development and Disease with Human Induced Pluripotent Stem Cells
Guo-Li Ming of the University of Pennsylvania is developing 3-dimensional cell culture models of the developing brain—so-called organoids—using induced pluripotent stem cells. High school students working in her lab designed 3D-printed lids with shafts that insert into standard cell culture plates, to divide each individual well of the plate into a separate miniaturized spinning bioreactor. Because most brain organoid protocols produced highly heterogeneous tissue, she used these tiny bioreactors to create organoids containing almost exclusively forebrain tissue.
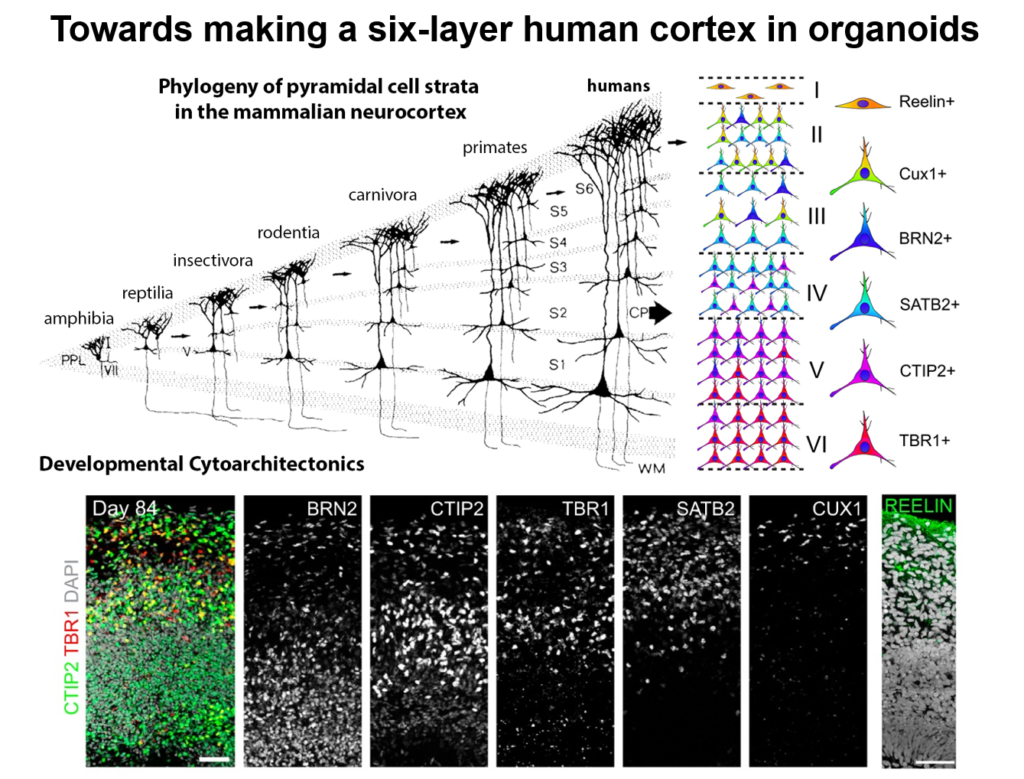
Cell labeling and gene expression studies showed that when grown for 100 days, these organoids recapitulated fetal forebrain development through the end of the second trimester. Progenitor cells generated neurons and glia whose migration pattern mirrored development, and the neurons received both excitatory and inhibitory input. The researchers used the organoids to study how Zika virus affects the developing brain. They found that the virus specifically targets neural progenitor cells, dose-dependently causing cell death and causing a collapse of tissue that resembles the microcephaly in infants affected by Zika. A screen of 6000 compounds yielded a neuroprotective compound called Emricasan that is positioned to enter clinical trials.
The group has now developed other brain-region specific organoids, modeling the midbrain and the hypothalamus. They plan to use these tools to study other neurodevelopmental disorders. Recent publications suggest the approach can also recapitulate features of neurodegenerative diseases, Ming said.
Engineering Neurogenesis via Lineage Reprogramming
For the past decade, Benedikit Berninger of Johannes Gutenberg University Mainz has been working on identifying cellular signals that can drive the reprogramming of astroglial cells from early postnatal mouse brain into neurons. More recently, to see if such reprogramming could be conducted in human cells, his lab began working with cells derived from adult human brains during epilepsy surgery. These cells turned out to be pericytes, and Berninger’s team identified a two transcription factors—Sox2 and Ascl1—that could reprogram them into functional neurons, which formed synapses and fired action potentials in culture.
To understand how the two transcription factors interact, the researchers investigated gene expression in the early stages—day 2 and day 7—in this two-week reprogramming process. A few genes were regulated by just one of the factors, but most were turned on only when both factors were present, suggesting that the two factors act synergistically. Ascl1 alone appears to target a different set of genes—ones associated with mesodermal cell fate (which generate pericytes), rather than neurogenesis-related genes activated when Ascl1 is co-expressed with Sox2. A similar difference was seen on a single cell level.
The researchers also observed two subpopulations in the starting population of pericytes—one of which was susceptible to reprogramming into neurons while the other was not. That may account for distinct competence in reprogramming in individual cells, Berninger said. For example, cells expressing the leptin receptor had a low level of reprogramming efficiency, indicating subtype differences in reprograming competence.
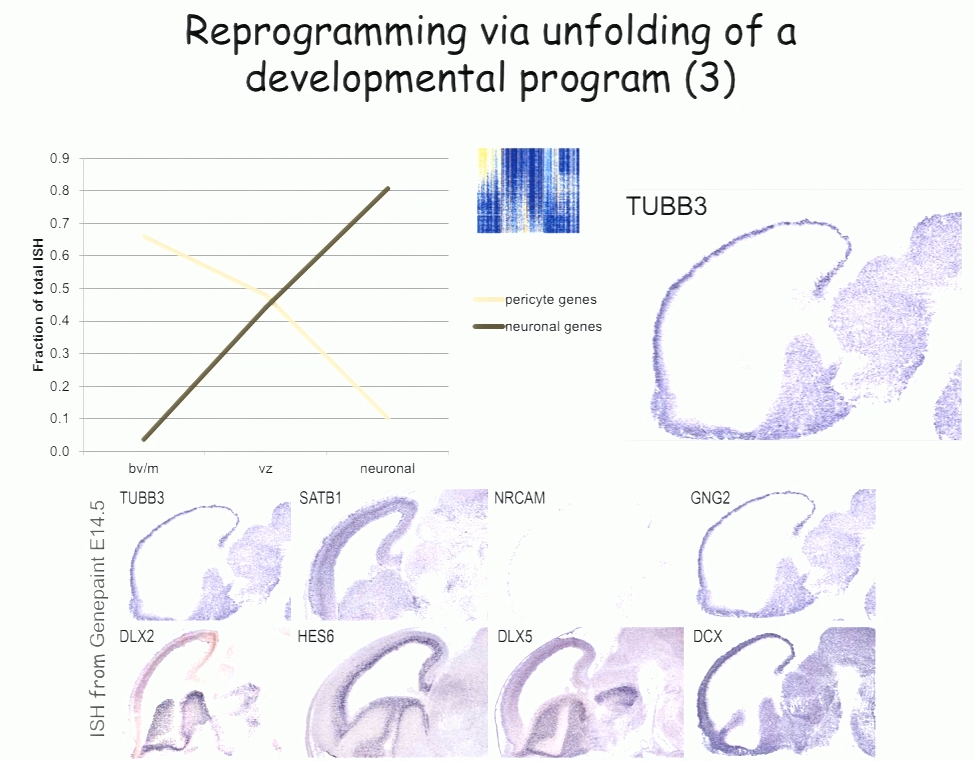
In the subset of cells that do reprogram successfully, a set of genes was induced transiently, then downregulated. These genes reflect a progenitor-like stage in the reprogramming process. These studies suggest that cells are not transforming directly from pericyte to neuron, but undergo a series of events reminiscent of an unfolding developmental program, Berninger said.
Rejuvenating and Re-engineering Aging Memory Circuits
The hippocampus plays a critical role in formation of episodic memories-that is, memories of what, when, and where. Essential to this capacity is the need to keep similar memories separate and retrieve past memories in a context appropriate manner. With age, the ability to keep similar memories separate and context-appropriate retrieval is potentially impaired, said Amar Sahay of Massachusetts General Hospital and Harvard Medical School. Within the hippocampus, the dentate gyrus-CA3 circuit performs operations such as pattern separation and pattern completion that support resolution of memory interference and retrieval. With age, neurogenesis in the hippocampus declines and CA3 neurons become hyper excitable in rodents, non-human primates and humans. Sahay’s lab investigates circuit mechanisms that may be harnessed to optimize hippocampal memory functions in adulthood and aging.
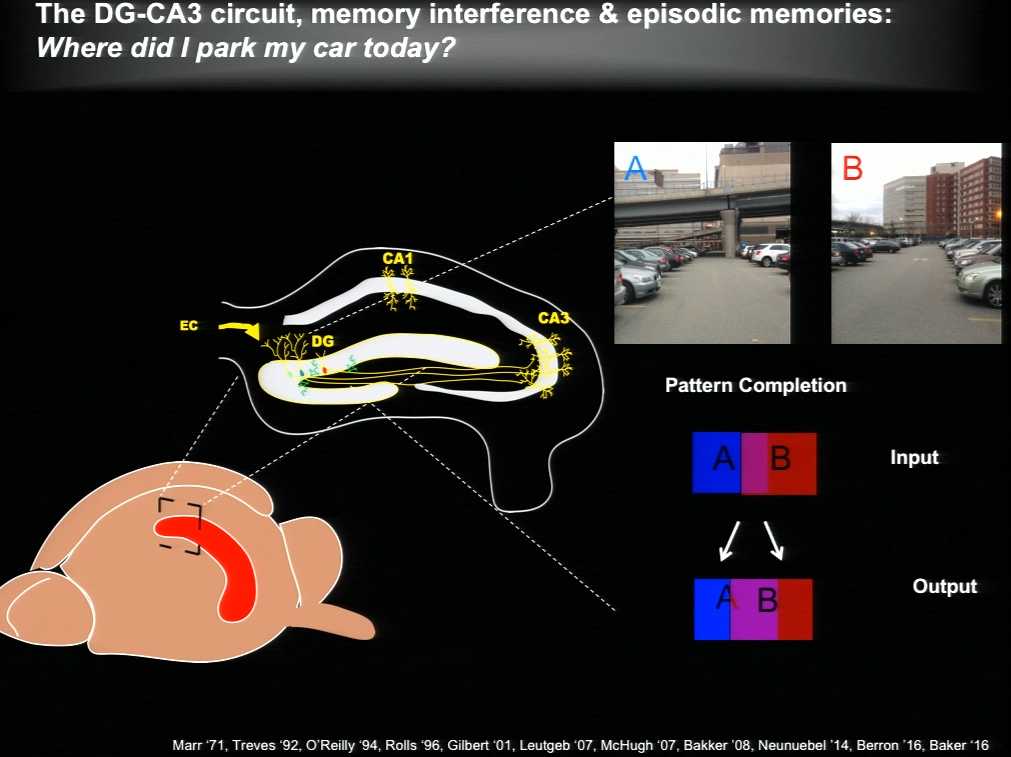
The hippocampus generates new neurons throughout life, and previous work has suggested that adult-born neurons integrate into the hippocampal circuitry by competing with existing mature neurons for inputs. Sahay and his colleagues identified a transcription factor called Klf9 that, when unregulated just in the mature neurons, biases competition dynamics in favor of integration of the adult-born neurons. This enhances neurogenesis in adult (3-month-old), middle-aged (12 months) and in aged (17-month-old) mice. Older rejuvenated animals (with enhanced adult hippocampal neurogenesis) had a memory advantage: they were better at discriminating between two similar contexts, one safe and one associated with a mild footshock.
In a complementary series of experiments, Sahay and his colleagues found age-related changes in connectivity between dentate granule neurons and inhibitory interneurons. They performed a screen and identified a factor with which they re-engineered connectivity between dentate granule neurons and inhibitory interneurons and augmented feed-forward inhibition onto CA3. By targeting this factor in the dentate gyrus of aged mice, the authors were able to reverse age-related alterations in dentate granule neuron-inhibitory interneuron connectivity and enhance memory precision.
Speaker Presentations
Further Readings
Graham Collingridge
Collingridge GL, Peineau S, Howland JG, Wang YT.
Long-term depression in the CNS.
Nat Rev Neurosci. 2010 Jul;11(7):459-73. doi: 10.1038/nrn2867. Review.
Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, Cho K.
Aβ(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β.
Nat Neurosci. 2011 May;14(5):545-7. doi: 10.1038/nn.2785. Epub 2011 Mar 27.
Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S, Brown C, Hashikawa T, Murayama M, Seok H, Sotiropoulos I, Kim E, Collingridge GL, Takashima A, Cho K.
Microtubule-associated protein tau is essential for long-term depression in the hippocampus.
Philos Trans R Soc Lond B Biol Sci.2013 Dec 2;369(1633):20130144. doi: 10.1098/rstb.2013.0144. Print 2014 Jan 5.
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T, Matthews P, Isaac JT, Bortolotto ZA, Wang YT, Collingridge GL.
LTP inhibits LTD in the hippocampus via regulation of GSK3beta.
Neuron. 2007 Mar 1;53(5):703-17.
Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M.
Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation.
J Neurosci. 2011 Feb 2;31(5):1688-92. doi: 10.1523/JNEUROSCI.2610-10.2011.
Guo-li Ming
Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, et al.
Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure.
Cell. 2016 May 19;165(5):1238-54. doi: 10.1016/j.cell.2016.04.032. Epub 2016 Apr 22.
Benedikit Berninger
Heinrich C, Blum R, Gascón S, Masserdotti G, Tripathi P, Sánchez R, Tiedt S, Schroeder T, Götz M, Berninger B.
Directing astroglia from the cerebral cortex into subtype specific functional neurons.
PLoS Biol. 2010 May 18;8(5):e1000373. doi: 10.1371/journal.pbio.1000373.
Karow M, Schichor C, Beckervordersandforth R, Berninger B.
Lineage-reprogramming of pericyte-derived cells of the adult human brain into induced neurons.
J Vis Exp. 2014 May 12;(87). doi: 10.3791/51433.
Karow M, Sánchez R, Schichor C, Masserdotti G, Ortega F, Heinrich C, Gascón S, Khan MA, Lie DC, Dellavalle A, Cossu G, Goldbrunner R, Götz M, Berninger B.
Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells.
Cell Stem Cell. 2012 Oct 5;11(4):471-6. doi: 10.1016/j.stem.2012.07.007.
Amar Sahay
McAvoy K, Besnard A, Sahay A.
Front Syst Neurosci. 2015 Aug 20;9:120. doi: 10.3389/fnsys.2015.00120.
McAvoy KM, Sahay A.
Targeting Adult Neurogenesis to Optimize Hippocampal Circuits in Aging.
Neurotherapeutics. 2017 Jul;14(3):630-645. doi: 10.1007/s13311-017-0539-6. Review.
McAvoy KM, Scobie KN, Berger S, Russo C, Guo N, Decharatanachart P, Vega-Ramirez H, Miake-Lye S, Whalen M, Nelson M, Bergami M, Bartsch D, Hen R, Berninger B, Sahay A.
Modulating Neuronal Competition Dynamics in the Dentate Gyrus to Rejuvenate Aging Memory Circuits.
Neuron. 2016 Sep 21;91(6):1356-73. doi: 10.1016/j.neuron.2016.08.009. Epub 2016 Sep 1.
Biomarkers, Hot Topics, and the Future of Therapeutics
Speakers
Reisa Sperling
Brigham and Women’s Hospital
Johanna Jackson
Eli Lilly and Company
Eliška Zlámalová
University of Cambridge
Arthur Konnerth
Technical University of Munich
Milo Robert Smith
Icahn School of Medicine at Mount Sinai
Highlights
- Multimodal imaging is becoming advanced enough to identify people with early-stage disease, which will help determine the critical window for therapies in clinical trials.
- Slow wave oscillations are disrupted in Alzheimer’s disease model mice due to a misregulation of excitatory and inhibitory synaptic activity.
- Imaging pre- and post-synaptic structures over time can reveal how disease progression affects synapses.
- Integrative bioinformatics can identify common pathways across neurodegenerative diseases and as well as drugs that can may act on those pathways.
- An RNAi-based screen in Drosophila can reveal genes that shape the morphology of axonal ER.
Neuroimaging in Early Alzheimer’s Disease
Alzheimer’s disease evolves over a couple decades, but most research to date has examined the disease at a late stage—perhaps too late to intervene effectively, said Reisa Sperling of Brigham and Women’s Hospital. Multimodal imaging is becoming advanced enough to identify people with early-stage disease, which will help determine the critical window for therapies in clinical trials.
PET amyloid imaging detects amyloid pathology in humans in vivo. Some 30% of clinically normal individuals have high amyloid levels, accumulating data suggests that this increases the risk of cognitive decline over the next 15 years—particularly when combined with markers of neurodegeneration such as decreased hippocampal volume. Still, Sperling said, “I see that as a glass half full—we’ve got 15 years to intervene.”
Committing something to memory requires activation of a brain region called the medial temporal lobe, where tau accumulates in AD. It also requires disabling the so-called default mode network (DMN), a brain circuit active when the brain is not engaged in a particular task. Amyloid accumulation disrupts the DMN, and disruptions also emerge in other networks and the specificity with which they signal.
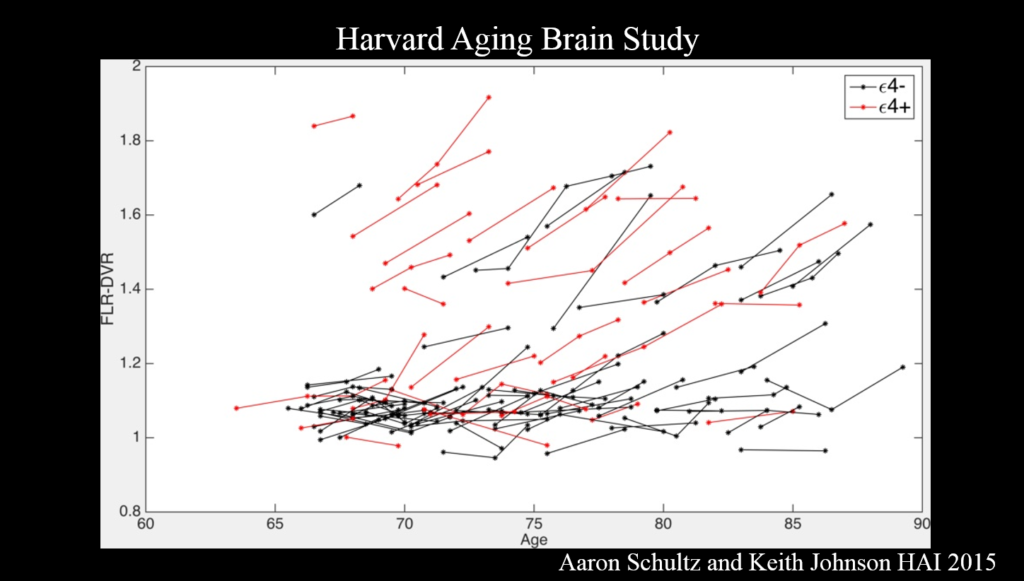
It’s the combination of amyloid and tau that is important for cognitive decline. Because tau—though not amyloid—correlates clearly with cognitive decline, tau PET imaging, which emerged just a couple years ago, has the most promise as a neurodegenerative marker for clinical trials, Sperling said. Ultimately, trials should move toward primary prevention—identifying drugs that block disease onset before clinical symptoms emerge. The field also needs biomarkers that show a person’s response to therapy.
Circuitry Dysfunction in Alzheimer’s Disease Mouse Models
A lot is known about clinical symptoms, pathology, and molecular mechanisms involved in Alzheimer’s disease, but there is a big gap in understanding how neuronal circuits are affected, said Arthur Konnerthof the Technical University of Munich.
About ten years ago, Konnerth’s lab developed a method for measuring neuronal function at the single cell level in living mice using fluorescent calcium indicators. They used it to investigate neurons surrounding amyloid beta plaques in mice lacking functional amyloid precursor protein (APP), an Alzheimer’s disease model. They hypothesized that these neurons would show decreased activity, but to their surprise, they were hyperactive, while further-away cells were silent. The error signal sent by these hyperactive cells probably disturbs the circuit significantly, Konnerth said.
His team also explored the function of long-range circuits in Alzheimer’s disease model mice. They studied slow wave oscillations, a form of activity that is essential for slow wave sleep and for memory consolidation. These waves travels through the cortex and into the hippocampus in a coherent fashion. In Alzheimer’s disease mice, the coherence of this circuitry is highly disrupted. Enhancing inhibitory (GABAergic) neuron activity reversed the deficit.
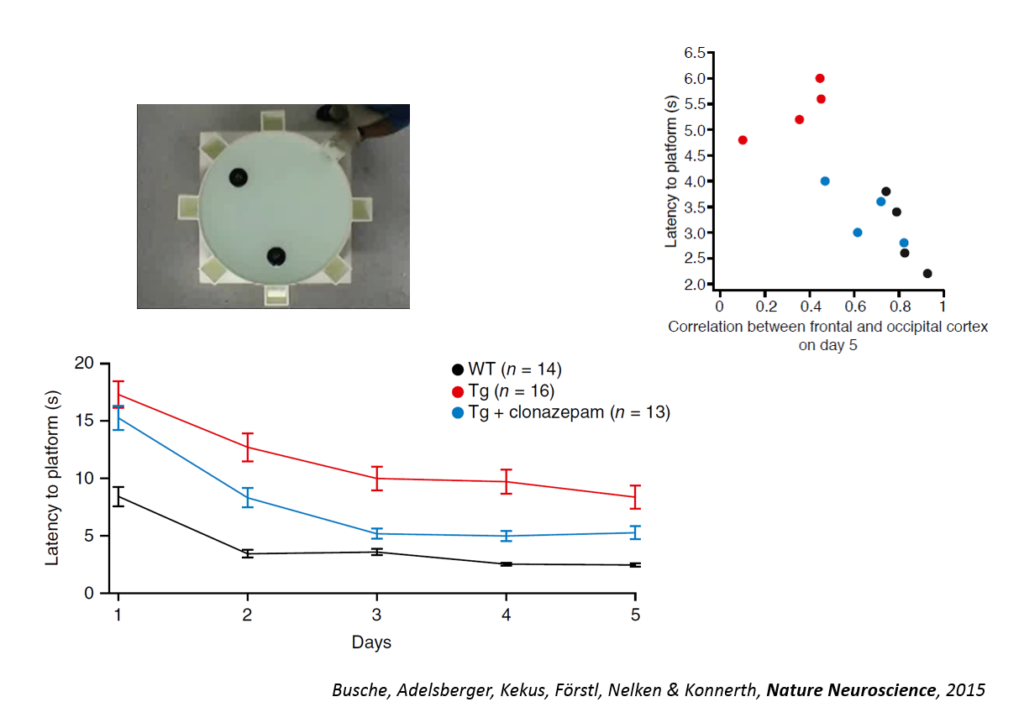
Tweaking GABAergic activity in normal mice also affected this circuitry, pointing to a synaptic effect. Returning the circuitry to normal also improved a learning task, the Morris water maze, and individual animals’ behavioral performance could be predicted by the coherence of this slow wave oscillation. An fMRI study in humans conducted by another lab showed also showed a disruption in slow wave oscillation. Targeting the shift in excitation-inhibition that underlies slow wave disruption may ameliorate cognitive deficits in the disease, Konnerth said.
Hot Topics in Neuroregeneration
In three short talks, early career researchers described imaging, bioinformatics and candidate gene analyses for probing neurodegenerative diseases.
Johanna Jackson from Eli Lilly used two-photon imaging in two mouse models of Alzheimer’s disease to study how disease progression affects synapses. She and her colleagues tracked axonal boutons and dendritic spines—the presynaptic and postsynaptic points of contact—over time in the same brain region. In the J20 mouse, which develops amyloid plaques, dendritic spine number remained constant, but axonal boutons were lost and the turnover rate of both spines and boutons increased as amyloidopathy progressed. The Tg4510s mouse, which develops tauopathy, showed a different pattern: both spines and boutons were lost, and neurites sickened then disappeared over time. Switching off the transgene in these mice could partially prevent or delay these deficits.
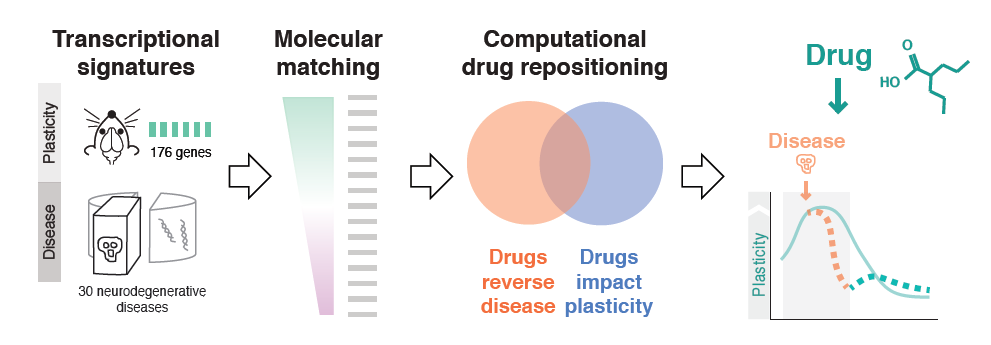
Milo Robert Smith of the Icahn School of Medicine at Mt. Sinai used bioinformatics to probe plasticity mechanisms in neurodegenerative diseases and to identify common disease pathways and potential therapeutic drugs. First, his team conducted microarray experiments to capture gene expression signatures of plasticity in mice. They then matched these signatures to transcriptomics signatures of 436 diseases taken from publicly available databases. The 100-plus illnesses showing a significant association were enriched for neurodegenerative diseases, and inflammatory genes appeared highly implicated. Finally, the researchers matched disease transcriptional signatures to transcriptional signatures of drugs measured in cell lines, also from publicly available databases. Using this approach, they identified drug candidates for resting plasticity in Huntington’s disease.
Human motor neuron axons can extend a meter in length, but dysfunction in trafficking such a distance underlies a neurodegenerative disease called hereditary spastic paraplegia (HSP), in which corticospinal motor neurons progressively degenerate. Eliška Zlámalová of the University of Cambridge is identifying candidate genes involved in long axon transport and HSP pathology. Three genes associated with HSP—reticulon, REEP1, and REEP2—produce proteins that localize to smooth endoplasmic reticulum (ER) in axons. When Zlámalová disabled all three in Dropsophila, ER fragmented in the middle of the axon and degenerated distally. To look for additional candidate genes, Zlámalová developed fluorescent markers for two other proteins, knocked own their genes in triple-mutant flies using RNA interference, and imaged ER morphology. She found a trend toward further ER fragmentation; a higher number of experiments may yield more conclusive results.
Further Readings
Reisa Sperling
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B.
A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers.
Neurology. 2016 Aug 2;87(5):539-47. doi: 10.1212/WNL.0000000000002923. Epub 2016 Jul 1. Review.
Schultz AP, Chhatwal JP, Hedden T, Mormino EC, Hanseeuw BJ, Sepulcre J, Huijbers W, LaPoint M, Buckley RF, Johnson KA, Sperling RA.
J Neurosci. 2017 Apr 19;37(16):4323-4331. doi: 10.1523/JNEUROSCI.3263-16.2017. Epub 2017 Mar 17.
Sperling R, Mormino E, Johnson K.
The evolution of preclinical Alzheimer’s disease: implications for prevention trials.
Neuron. 2014 Nov 5;84(3):608-22. doi: 10.1016/j.neuron.2014.10.038. Epub 2014 Nov 5. Review.
Sperling RA, Jack CR Jr, Aisen PS.
Testing the right target and right drug at the right stage.
Sci Transl Med. 2011 Nov 30;3(111):111cm33. doi: 10.1126/scitranslmed.3002609. Review.
Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA.
Neuron. 2009 Jul 30;63(2):178-88. doi: 10.1016/j.neuron.2009.07.003.
Arthur Konnerth
Busche MA, Kekuš M, Adelsberger H, Noda T, Förstl H, Nelken I, Konnerth A.
Rescue of long-range circuit dysfunction in Alzheimer’s disease models.
Nat Neurosci. 2015 Nov;18(11):1623-30. doi: 10.1038/nn.4137. Epub 2015 Oct 12.
Busche MA, Konnerth A.
Neuronal hyperactivity — a key defect in Alzheimer’s disease?
Bioessays. 2015 Jun;37(6):624-32. doi: 10.1002/bies.201500004. Epub 2015 Mar 14.
Busche MA, Grienberger C, Keskin AD, Song B, Neumann U, Staufenbiel M, Förstl
H, Konnerth A.
Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer’s models.
Nat Neurosci. 2015 Dec;18(12):1725-7. doi: 10.1038/nn.4163. Epub 2015 Nov 9.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O.
Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease.
Science. 2008 Sep 19;321(5896):1686-9. doi: 10.1126/science.1162844.
Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, et al.
η-Secretase processing of APP inhibits neuronal activity in the hippocampus.
Nature. 2015 Oct 15;526(7573):443-7. doi: 10.1038/nature14864. Epub 2015 Aug 31.
Johanna Jackson
Scheff SW, Price DA, Schmitt FA, Mufson EJ.
Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment.
Neurobiol Aging. 2006 Oct;27(10):1372-84. Epub 2005 Nov 9.
Milo Robert Smith
Smith MR, Burman P, Sadahiro M, Kidd BA, Dudley JT, Morishita H.
eNeuro. 2017 Jan 18;3(6). pii: ENEURO.0240-16.2016. doi: 10.1523/ENEURO.0240-16.2016. eCollection 2016 Nov-Dec.
Eliška Zlámalová
Blackstone C.
Cellular pathways of hereditary spastic paraplegia.
Annu Rev Neurosci. 2012;35:25-47. doi: 10.1146/annurev-neuro-062111-150400. Epub 2012 Apr 20.
Gerondopoulos A, Bastos RN, Yoshimura S, Anderson R, Carpanini S, Aligianis I, Handley MT, Barr FA.
Rab18 and a Rab18 GEF complex are required for normal ER structure.
J Cell Biol. 2014 Jun 9;205(5):707-20. doi: 10.1083/jcb.201403026. Epub 2014 Jun 2.
Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, et al.
Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders.
Science. 2014 Jan 31;343(6170):506-511. doi: 10.1126/science.1247363.
Panel Discussion: The Future of Research and Therapies in Neuroregeneration and Restoration
Speakers
Michael J. O’Neill, Moderator
Eli Lilly and Company
Ana Maria Cuervo, Panelist
Albert Einstein College of Medicine
Mark P. Mattson, Panelist
National Institute of Aging
Clive Svendsen, Panelist
Cedars-Sinai Medical Center
Jeffrey Macklis, Panelist
Harvard University
The panelists began by summarizing what they consider the most exciting dimension in the field of regeneration. Jeffery Macklis said that since graduate school, he had puzzled over the fact that only certain cell types were vulnerable and selectively damaged in different neurodegenerative diseases. “I find that the most exciting question,” he said. “Until we get to neuron subtype specificity and the circuits involved, we could be looking at a lot of unrelated stuff.”
Ana Maria Cuervo notes that neurodegenerative diseases primarily occur in the elder population, yet researchers still don’t know enough about the physiology of aging to determine which dimensions of the disease are due to aging and which are not.
Mark P. Mattson agreed, noting that in Alzheimer’s disease, events upstream of amyloid including generic age-related events such as increased oxidative stress, can affect the disease. “We need to understand those if we want to intervene earlier,” he said. He also wondered whether mechanisms being targeted by drug development could also be activated by exercise or energy restriction. A related approach might be to induce mild intermittent bioenergetic stress on cell pharmacology.
“The thing that keeps me up at night in this field is biomarkers,” said Clive Svendsen. Molecules that change as the disease progresses are not necessarily causative; indeed, some of the stress responses observed in Alzheimer’s disease might be neuroprotective, and that holds for Huntigton’s disease, too, he explained.
An audience member raised the question of sex differences in neurodegenerative disease, noting that even when boys and girls reach the same cognitive milestones, they often arrive there through different routes. In response, Mattson described a study conducted by his group that compared responses to different diets in male versus female mice. At 40% calorie restriction, females shut down their estrus cycle, increased their physical activity, and lost most of their body fat. Males under the same circumstances remained fertile, and their activity levels did not change. That could be because from an evolutionary perspective, females would ostensibly want to avoid having babies when there is no food around, because they lack the energy to care for them, while males might want to inseminate as widely as possible before they starve to death.
Reisa Sperling noted that women respond more adversely to a smaller amount of amyloid beta. “Something about being female means that you are more vulnerable,” she noted. An audience member noted that although men have a higher risk of Parkinson’s disease, females deteriorate faster once diagnosed. Svendsen noted that these observations speak to broader issues in personalizing treatments for neurodegenerative diseases. Sporadic Alzheimer’s disease likely consists of more than one disease, for example. “We’re trying to subdivide ALS into 10 types,” he said.
Panel Discussion
Open Questions
- How do hypertrophic astrocytes help require damaged neuronal circuits?
- What is the best way of clinically testing the amyloid beta hypothesis?
- Can the signaling mechanism linking neuronal activity at the synapse and gene transcription in the nucleus be therapeutically targeted?
- How should Parkinson’s disease therapeutic efforts account for homeostatic plasticity in stratal neurons?
- Why do different inflammatory responses have different effects on CNS regeneration? [Giger]
- How can growth cone machinery be targeted to promote regeneration?
- Can fasting and exercise mitigate against dementia and neurodegenerative damage in diseases like Alzheimer’s and Parkinson’s?
- How do pathogenic proteins cause the breakdown of chaperone-mediated autophagy, and how does such authophagy contribute to the clearance of pathogenic proteins?
- Will improvements in mitochondrial function obtained by targeting Parkin, PARIS or related proteins provide therapeutic benefits in Parkinson’s disease?
- How does glial cell dysfunction cause neurological disease and can it be therapeutically targeted? [Goldman]
- Can a cell therapy consisting of GDNF-releasing astrocytes stave off paralysis in ALS?
- Are there neurodegenerative diseases besides ALS in which genes are maladaptively downregulated in astrocytes?
- Will drugs that modulate NMDA activity prove beneficial for Alzheimer’s disease?
- How well can organoids reflect the pathology of neurodegenerative diseases?
- Can promoting reprogramming strategies that turn non-neuronal cells into neurons be used therapeutically?
- Can memory be improved with the help of molecular strategies to rejuvenate hippocampal circuitry that degenerates with age?
- Will candidates identified through integrative bioinformatics yield drugs that target common mechanisms in neurodegenerative disease?
- How to determine the optimal window for efficacy of different prospective Alzheimer’s disease therapies?
- Will reversing the disintegration of slow wave oscillations ameliorate cognitive impairment in Alzheimer’s disease?