A panel of experts from across sectors discuss possible applications and open questions.
Published November 20, 2018
By Marie Gentile, Mandy Carr, and Richard Birchard
From your smartphone to personal computers. From at-home genetic tests to insurance databases. There is a tremendous amount of data out there that relates to our health. Not all of it is being used yet by those who help manage our healthcare, but it’s only a matter of time before that changes. What influences will this and other data have on our health and the healthcare system at large?
In this video, you’ll hear from Jacqueline Corrigan-Curay, JD, MD (U.S. Food and Drug Administration), Brett Davis (Deloitte), Vivian Lee, MD, PhD, MBA (Verily), and Patrick Ryan, PhD (Janssen & Columbia University), with moderation from Mark Sheehan, PhD (The Ethox Centre, University of Oxford).
They spoke in the first panel at “Healthcare in the Era of Big Data: Opportunities and Challenges,” a collaboration with New York University. This 2-day symposium explored the ethical risks and rewards of incorporating big data into the healthcare landscape.
View other talks and panels from the symposium on our Livestream channel.
Ellie Zillfleisch looks forward to the day where she might help others suffering from Chronic Recurrent Multifocal Osteomyelitis.
Published October 22, 2018
By Marie Gentile, Mandy Carr, and Richard Birchard
A hospital bed might not be where you’d expect to find a career revelation, but that’s where Ellie Zillfleisch, 14, discovered her love for STEM. She grew up in Julatten, a small, rural town in Queensland, Australia, home to just 1,000 people. At 11, doctors diagnosed her with Chronic Recurrent Multifocal Osteomyelitis (CRMO), a disease that develops bone lesions. CRMO affects 1 out of every 1,000,000 people.
“My bones look like honeycombs, which is kind of cool (even though it’s painful),” says Ellie.
There is no standard treatment for CRMO. She started having symptoms when she was eight, and doctors routinely misdiagnosed her with rheumatoid arthritis, cancer, and osteomyelitis. Going to hospitals in big cities intimidated Ellie, who was used to her small town life.
A First Foray into Medicine
After spending a month in a hospital in Brisbane, she started having acute anxiety attacks. Her fear of needles grew when she thought her IVs would fall out. To prove they’d stay in place, doctors let her take off the tape that held the tubes in place. Ellie thinks of this as her first foray into medicine.
To overcome CRMO, Ellie found inspiration from the superhero, Green Arrow, whose superpower involves using trick arrows to stop bad things from happening and who often refers to this Russian proverb: “the shark that doesn’t swim drowns.”
“If I did not beat this disease, it would swallow me,” she told us. “I often thank those doctors in the hospital all those years ago, as now I am hoping to pursue medicine as a career and say, ‘I shattered this disease.’”
Ellie Zillfleisch met her mentor, Courtney Veilleux, at the GSA Summit.
Chasing a Dream
Despite her chronic disease and small-town roots, she looks for every opportunity to get closer to her dream. Ellie heard about The New York Academy of Sciences’ 1000 Girls, 1000 Futures program from a friend who took part. When she realized a STEM mentor could give her the edge in college and her future career, she applied immediately.
Ellie felt overwhelmed when she started 1000 Girls, 1000 Futures. She wasn’t sure if she would have enough time to participate while staying on track with school and other extracurricular activities. Her mentor reassured her she was capable of completing all her tasks and taught her to balance her busy schedule. Ellie believes she improved her work-life balance by setting manageable goals for each day.
One of the opportunities 1000 Girls, 1000 Futures provided her was attending the Global STEM Alliance Summit in New York City. She received an all-expense paid trip to New York because she was picked as a “Mentee of the Month.” Mentors nominate students for this award for being active and exemplary participants.
Interacting with a global community of students has shown Ellie a world outside her own in Julatten. She even wants to attend college in the United States because she believes there are more opportunities for women in STEM there.
As a board-certified pediatrician with a decade and a half of experience, Dr. Amanda Dempsey shares strategies for discussing vaccines with skeptical parents.
Published September 11, 2018
By Andre Legaspi
The emergence of a powerful anti-vaccination movement is now threatening the progress made by vaccines in the 20th century. Amanda Dempsey, MD, PhD, MPH is a pediatrician confronting the myths surrounding vaccines through innovative communication techniques, with exciting results.
Why is there an increasing skepticism regarding vaccines?
It begins with the media environment. Information is now much more accessible than it was a couple decades ago. Contrary opinions can have a strong voice in social media, and the internet, and can spread just as easily as more reliable sources of information. This leads people to believe that there are two equal sides, even though there’s a lot more scientific evidence supporting the necessity and the safety of vaccines than there is about their potential downsides.
What do you tell a parent who is skeptical about immunization?
I would ask about the underlying reasons for their skepticism. Honing in on one specific concern is important because you don’t want to inadvertently raise new concerns for that parent.
Then I would focus on the fact that vaccination is the norm, and I would explain why vaccination is important. I cover the safety process that vaccines undergo, the vaccination risks—which are minimal, but not zero—and finally make the case for why the risks associated with vaccinations are outweighed by the benefits.
Do you have a method for dealing with parents who remain unconvinced?
Amanda Dempsey, MD, PhD, MPH
One technique is motivational interviewing. This communication style was initially developed for helping healthcare providers address motivation and compliance issues in patients with substance abuse.
My group adapted some tools from motivational interviewing and applied them to our conversations about vaccines with parents who have questions or concerns. It changes the dynamic of the patient-provider relationship. I’m no longer on the opposite side of the vaccination issue, I’m helping them think through their decision-making in a way that feels like we’re working towards a joint decision.
Research shows that taking a paternalistic approach—just saying vaccines are the norm—is effective, but only for parents who are mostly, or completely on board with vaccines. For the parents that have more substantial concerns or questions, simplistic approaches aren’t going to work. That’s where turning to more nuanced techniques, like motivational interviewing, can be quite helpful.
What is the Ready Vac study?
The Ready Vac study is taking a different approach, called tailored messaging. We try to focus the conversation on the concerns that a parent actually has, so as not to overwhelm them with too much information, which can cause some people to tune out. We want to highlight the information that most speaks to their particular concerns or needs.
The need for this type of intervention arises from the fact that when parents have a lot of questions about vaccines, there’s not enough time in a regular clinical visit to be able to discuss all of their concerns.
How will this study help promote vaccinations?
We created an online tool that customizes information about vaccines based on each parent’s unique values, attitudes, beliefs, and experiences. Each person gets a different compilation of information depending on their major concerns about the vaccines.
This way, you’re providing relevant information to that person and not diluting the impact with extraneous information that they don’t care about or want to know.
The hope is that this sort of automated tool will be effective at improving parents’ vaccination attitudes, acceptance, and ultimately the rate at which their children are up to date with recommended vaccines.
For centuries, physicians and scientists have thought of inflammation as the body’s acute response to infection or injury, but in recent decades it’s become clear that chronic inflammation drives pathologies as diverse as cancer, diabetes, and Alzheimer’s disease. Controlling this aberrant inflammation, however, has proven difficult. Conventional anti-inflammatory drugs work by antagonizing the body’s pro-inflammatory hormones, but that approach also suppresses immunity, opening the patient to secondary infections. A newer strategy relies on recently discovered resolution mediators, compounds that the body makes naturally to resolve inflammatory responses without suppressing other parts of the immune system. Drugs targeting this process have shown immense potential to treat many of the world’s most serious diseases, with fewer side effects than existing therapies.
On June 25–26, 2018, the New York Academy of Sciences hosted Resolution of Inflammation, Infection and Tissue Regeneration, a symposium featuring many of the top researchers in the rapidly developing field of resolution pharmacology. In two days of oral presentations, a poster session, and an extensive panel discussion, speakers and attendees reviewed the biggest advances and challenges in resolution biology. The meeting covered the basic biology of inflammation and its resolution, studies on animal models of chronic and acute diseases, and clinical trials of promising new inflammation-resolving drugs.
Speakers
Nan Chiang, PhD Brigham and Women’s Hospital, Harvard Medical School
Michael Conte, MD University of California, San Francisco
Jesmond Dalli, PhD William Harvey Research Institute, QMUL
Gabrielle Fredman, PhD Albany Medical Center
Catherine Godson, PhD University College Dublin
Dipak Panigrahy, MD Beth Israel Deaconess Medical Center, Harvard Medical Center
Paul Ridker, MD, MPH Brigham and Women’s Hospital, Harvard Medical School
Charles Serhan, PhD, DSc Brigham and Women’s Hospital, Harvard Medical School
Patricia Sime, MD University of Rochester School of Medicine
Matthew Spite, PhD Brigham and Women’s Hospital, Harvard Medical School
Ira Tabas, MD, PhD Columbia University
Mark Tepper, PhD Corbus Pharmaceuticals
Kevin Tracey, MD The Feinstein Institute for Medical Research
Thomas Van Dyke, DDS, PhD Forsyth Center for Clinical and Translational Research
Resolution Mediators and Mechanisms in Inflammation: Leads for 21st Century
Speakers
Charles Serhan Brigham and Women’s Hospital, Harvard Medical School
Charles Serhan, from the Brigham and Women’s Hospital and Harvard Medical School, opened the meeting with a keynote presentation that spanned the history of resolution physiology, a field he pioneered. Physicians and scientists have known about inflammation since antiquity, primarily as an acute condition associated with injury and infection. In recent years, however, biologists have come to understand that chronic inflammation underlies many non-communicable diseases, including cancer, diabetes, Alzheimer’s disease, and Parkinson’s disease.
Serhan has focused on how acute inflammation normally resolves, and how this process sometimes malfunctions, leading to chronic inflammation. When he entered the field, “in the textbooks, the resolution of acute inflammation was thought to be a passive event,” Serhan said. Pro-inflammatory molecules became diluted over time, bringing the inflammation to an end. Through an extensive series of cell culture, animal, and human studies, Serhan and his colleagues have overturned that model, showing that multiple families of pro-resolving molecules actively antagonize the inflammatory process and promote healing. These small fatty acid-derived molecules, now known as lipoxins, resolvins, protectins, and maresins, act through specific cellular receptors to orchestrate a complex switch from inflammation to resolution.
Several classes of chemical mediators actively drive inflammation resolution.
Serhan, like many others at the meeting, is now exploiting those findings to design new drugs that could treat a wide range of chronic conditions far more effectively than current anti-inflammatory compounds, with fewer side effects. “This is really a paradigm shift in thinking about how to treat inflammation, using agonists to stimulate resolution rather than inhibitors that eventually become immunosuppressive,” said Serhan.
Dipak Panigrahy, MD Beth Israel Deaconess Medical Center, Harvard Medical Center
Ira Tabas, MD, PhD Columbia University
Cancer Progression: Failure to Resolve?
Dipak Panigrahy, of Beth Israel Deaconess Medical Center and Harvard Medical School, started the meeting’s first session by discussing his work on inflammation resolution in cancer. Researchers first discovered the link between inflammation and tumor growth in the late 19th century, but only recently has it become clear that inflammation is an essential initiator of at least some types of cancer. A mouse model of pancreatic cancer, for example, does not get the disease unless it first develops pancreatitis.
Unfortunately, most current anti-inflammatory drugs suppress the immune system, allowing established tumors to become even more aggressive. To find better solutions, Panigrahy began dissecting the mechanisms linking cancer and inflammation. He discovered that cancer-killing interventions such as chemotherapy leave behind debris from the killed tumor cells. In a mouse model of ovarian cancer, that debris worsens the disease. “So the debris will stimulate the tumor growth. It’s kind of a double edged sword of cancer therapy,” said Panigrahy. Adding specialized pro-resolving mediator molecules (SPMs) stimulates phagocytic cells to engulf the cellular debris, inhibiting further tumor growth. Treatments that boost SPMs could work synergistically with chemotherapy to attack numerous types of cancer.
The Interplay between Efferocytosis and Inflammation Resolution
Ira Tabas from Columbia University continued the theme of clearing cellular debris, with a presentation about his group’s work on the process. Macrophage cells function as the body’s garbage collectors, engulfing the remains of dead cells through efferocytosis. The process is critical for healing damaged tissue and resolving inflammation, but it requires intensive metabolic management by the macrophage. “When a macrophage is eating multiple dead cells … it might be like you sitting down eating 20 filet mignons, and then half an hour later eating 20 more,” said Tabas, adding that “it’s a tremendous metabolic load.”
Efferocytosis of dead cells requires macrophages to handle a high metabolic load.
Tabas’s team studies efferocytosis in mouse models of heart disease. They found that eliminating a single gene in the animals blocks efferocytosis and leads to much worse disease, highlighting the importance of efferocytosis in resolving inflammation. Next, the researchers used an elegant in vitro assay to demonstrate that the degradation products from engulfing one dead cell regulate the macrophage’s ability to engulf a second one. The macrophages use another tightly regulated process to recycle the membranes of the dead cells. Together, the mechanisms explain how macrophages can keep their efferocytosis rates as high as possible without becoming metabolically overloaded.
Michael Conte, MD University of California, San Francisco
Gabrielle Fredman, PhD Albany Medical Center
Paul Ridker, MD, MPH Brigham and Women’s Hospital, Harvard Medical School
Providing Proof of Principle for Atherosclerosis, Lung Cancer, Kidney Disease, and Osteoarthritis: Lessons from CANTOS
Paul Ridker from Brigham and Women’s Hospital began the second session with a keynote address on the CANTOS clinical trial. This trial sought to address several questions about inflammation resolution in cardiovascular disease.
Atherosclerosis is a leading cause of vascular disease worldwide. Researchers have identified numerous factors that predict the onset of atherosclerosis, including high blood pressure, cholesterol, and markers of chronic inflammation, but it remains unclear which of these factors are causative and which merely correlate with pathogenesis.
Current treatments for atherosclerosis focus on lowering blood pressure and cholesterol levels, but Ridker and his colleagues wanted to target inflammation instead. To do that, they used a human monoclonal antibody drug called canakinumab, which binds the pro-inflammatory protein interleukin-1beta. IL-1beta triggers a series of signals that promote inflammation and also increase blood levels of C-reactive protein (CRP), a biomarker of atherosclerosis. A pilot study on 1,400 patients sought to determine how much canakinumab it would take to reduce patients’ CRP levels to baseline, but the drug was so powerful that it worked at all doses tested.
The CANTOS trial enrolled over 10,000 patients to test canakinumab in patients with cardiovascular disease.
Based on those data, Ridker’s team designed a much larger trial, enrolling over 10,000 patients. The participants all had high CRP levels even after aggressive cholesterol-lowering therapy, indicating that they were still suffering from chronic inflammation. Those treated with canakinumab showed dramatic reductions in CRP, and concomitantly lower rates of cardiovascular events over the trial’s seven-year run, compared to controls who received a placebo. Crucially, treated patients showed no signs of immunosuppression, and most of the drug’s side effects were mild or even beneficial. For example, patients in the treatment groups had significantly lower rates of overall cancer mortality and chronic kidney disease than controls.
The results show that atherosclerosis treatment plans should focus on both cholesterol and inflammation. “If we’re going to beat this disease, we have to [target] both of these processes,” said Ridker.
Resolution of Vascular Injury: Mechanisms and Therapeutic Implications
Michael Conte, of the University of California, San Francisco, addressed what happens in the next step of modern vascular disease management: surgery. “It really doesn’t matter what we do when we touch blood vessels, whether we inflate a balloon, scrape out plaque, do a bypass graft or insert a catheter, we are faced with a scarring response … that has a classic inflammatory and resolution phase,” Conte explained.
In animal models of vascular injury, damaged blood vessels become inflamed, then produce specialized pro-resolving mediators (SPMs) to resolve the inflammation. Conte’s group also found that delivering extra SPMs locally to the site of injury significantly improves inflammation resolution and healing.
To put that finding into practice, the investigators have developed thin film polymers that can release SPMs directly into a vessel over time. They wrap the film around the blood vessel during surgery and leave it in place to deliver the molecules for days afterward. The system reduces graft thickness in a rabbit bypass graft model, and improves outcomes in mouse models of aneurysm surgery and thrombosis. Conte is also conducting a clinical trial testing the safety of naturally isolated SPM-stimulating compounds in humans.
Dysregulation of Resolution Pathways in Atherosclerosis
Gabrielle Fredman from Albany Medical Center continued the vascular theme, discussing her work on how dead cells get removed from atherosclerotic plaques. As a plaque develops on the wall of an artery, the cells at the center begin to die. This weakens the structure of the plaque and makes it more likely to rupture. Fredman studies this process in genetically modified mice that develop atherosclerosis when fed a high-fat diet. Treating these mice with SPMs decreases necrosis in their plaques, but how?
When cells die through apoptosis, or programmed suicide, macrophages rapidly engulf the resulting debris. Fredman and her colleagues found that cells undergoing necroptosis, such as those in the core of a plaque, are apparently much less appetizing. Macrophages fed necroptotic cells take them up much more slowly and in smaller pieces. “This resembles something like a nibbling process rather than a whole engulfment process,” said Fredman. The researchers subsequently found that a molecule called CD47 on the necroptotic cells may act as a “don’t eat me” signal, slowing their engulfment by macrophages. Adding SPMs, however, boosts macrophages’ efferocytosis responses enough to overcome that signal.
Speaker Presentations
Further Readings
Ridker
Aday, A.W., Lawler, P.R., Cook, N.R., et al. (2018).
Nan Chiang, PhD Brigham and Women’s Hospital, Harvard Medical School
Jesmond Dalli, PhD William Harvey Research Institute, QMUL
Kevin Tracey, MD The Feinstein Institute for Medical Research
The Role of n-3 Docosapentaneoic Acid-derived Pro-resolving Mediators in Systemic Protection
Jesmond Dalli, of the William Harvey Research Institute at the Queen Mary University of London, began the second part of the session with a presentation linking circadian rhythms and vascular inflammation. Disruption in circadian rhythms has been linked to metabolic disorders and cardiovascular disease progression, so Dalli wondered whether specialized pro-resolving mediators (SPMs) also showed circadian patterns.
Dalli’s team took blood samples from seven healthy volunteers throughout a 24-hour period, and found that the level of n-3 DPA, a precursor to SPMs, rose and fell in a consistent cycle. In contrast, patients with cardiovascular disease lose this circadian regulation, and have consistently low levels of SPMs. Additional experiments in mice confirmed the link. “This suggests that [loss] in circadian regulation of this pathway leads to a breakdown [in] early morning activation of platelets and monocytes,” said Dalli, “and that this then promotes cardiovascular disease.” He and his colleagues found that the cholesterol-lowering drugs atorvastatin and pravastatin increase the production of two SPMs, reinforcing other groups’ findings that these drugs combat cardiovascular disease through multiple mechanisms.
Pro-resolving Receptors: Mechanisms and Signaling
Nan Chiang from Harvard Medical School talked about the cellular receptors that respond to SPMs. One well-characterized SPM, resolvin D2, is a potent promoter of inflammation resolution; even extremely low doses of resolvin D2 can limit peritonitis and enhance survival in animal models of sepsis.
Resolvin molecules signal through specific cellular receptors to generate their observed effects.
Chiang suspected that resolvin D2 might act through a G-protein coupled receptor, so she tested a panel of 77 of these receptors whose functions had not yet been identified. That screen revealed that the receptor GPR18 responds strongly to resolvin D2. Human macrophages overexpressing GPR18 respond more strongly to resolvin D2 than controls; and mice lacking GPR18 don’t benefit from resolvin D2 treatment during experimental E. coli infection or sepsis, confirming that GPR18 is a biologically relevant receptor for resolvin D2.
In addition to resolvins, Chiang also studies another family of SPMs, known as maresin conjugates in tissue regeneration (MCTRs). She discovered that MCTRs act through a receptor called CysLT1 to reduce vascular leakage after injury. Knowing the receptors for these SPMs should facilitate the design of drugs with similar effects.
Molecular Approaches to Bioelectronic Medicine
Kevin Tracey, from the Feinstein Institute for Medical Research, started the meeting’s second day with a keynote presentation on bioelectronic medicine. Instead of manipulating body chemistry with chemicals, bioelectronic treatments connect electronic devices to the nervous system to stimulate desired responses. “The idea of bioelectronic medicine is a mechanistic approach to using electrons to replace drugs,” Tracey explained.
The vagus nerve connects the brain to multiple organs, and also the immune system.
About thirty years ago, when Tracey was a practicing neurosurgeon, a young patient died in his arms from shock. That spurred him to search for new drugs that could block shock-causing cytokines. In one experiment, his lab injected a promising anti-inflammatory drug directly into the brains of mice with experimentally induced strokes. It blocked inflammatory cytokines in the brain, but also in other organs throughout the animals’ bodies. That made no sense, as the molecule shouldn’t have been able to escape the brain.
The researchers subsequently discovered that the vagus nerve can trigger an anti-inflammatory reflex throughout the body, connecting the nervous system directly to the immune system. Subsequent work by others revealed that the vagus nerve stimulates the production of pro-resolving mediators in multiple organs and tissues. Tracey’s team is now conducting clinical trials to treat Crohn’s disease with an implanted electronic device that stimulates the vagus nerve and triggers this response.
Speaker Presentations
Further Readings
Dalli
Arnardottir, H., Orr, S.K., Dalli, J., and Serhan, C.N.
Proc. Natl. Acad. Sci. U.S.A. 2018; 115(21):E4843-E4852.
Session 3: Better Living through Chemistry
Speakers
Patricia Sime, MD University of Rochester School of Medicine
Matthew Spite, PhD Brigham and Women’s Hospital, Harvard Medical School
Temporal Biosynthesis of Pro-Resolving Lipid Mediators by Distinct Immune Cell Subsets during Skeletal Muscle Injury and Regeneration
Matthew Spite, of Brigham and Women’s Hospital and Harvard Medical School, returned the focus to chemistry, with a discussion of the specialized pro-resolving mediators (SPMs) that orchestrate healing from injuries. Using mass spectrometry, Spite identified all of the lipids produced in an injured mouse muscle as it recovers. The lipid profile changes over time and increases in the well-known resolvin subset of SPMs correlate with healing. That correlation persists whether the muscle damage comes from a toxin or exercise. “A lot of these pathways were consistent in two distinct models of injury,” said Spite. His team next characterized the macrophages present in the healing tissue, and found a distinct shift from pro-inflammatory to pro-resolving macrophage types, in sync with the shift in the lipid profile.
COPD: Inflammation, Infection and Resolution
Patricia Sime, from the University of Rochester School of Medicine, discussed the role of SPMs in chronic obstructive pulmonary disease (COPD). COPD, a chronic inflammatory condition, is now the fourth leading cause of death worldwide. It is caused primarily by smoking. “There’s actually no drug treatment for COPD,” said Sime, “so it’s a huge unmet need.”
Exposing mice to precisely controlled doses of cigarette smoke provides a model of human lung inflammation.
To better understand COPD, Sime used mass spectrometry to profile the lipids in exhaled breath condensates from patients with the disease as well as healthy controls. That pointed toward the SPMs resolvin D1 and D2 as critical mediators of inflammation resolution in the lung. Treating a mouse model of chronic smoking with resolvins decreases inflammation in the animals’ lungs. Sime hopes to exploit these findings to develop new therapies for COPD, as well as prophylaxis against lung damage from all types of smoke inhalation.
Thomas Van Dyke, DDS, PhD Forsyth Center for Clinical and Translational Research
Lipoxins and Lipoxin Mimetics Attenuate Diabetic Complications
Catherine Godson of University College Dublin opened the meeting’s final session with a presentation on lipoxins in diabetes. As a result of the obesity pandemic, diabetes has become an immense global health problem. One of the most serious complications of diabetes is kidney disease. “Those with the most profound diabetic kidney disease… have almost 50% increased mortality,” said Godson, adding that “there are no effective therapeutics” for the condition.
In an effort to change that, Godson and her colleagues have examined the lipoxin A4 molecule, a specialized pro-resolving molecule (SPM) that attenuates inflammation in animal models. The researchers found that lipoxin A4 attenuates molecular signals that promote fibrosis, helps maintain epithelial tissue integrity, and reduces activation of the inflammatory cytokine TGF-beta. “All of these are important potential targets of lipoxin actions in the context of chronic kidney disease and diabetic kidney disease,” said Godson.
Lipoxins mediate multiple protective and inflammation-resolving effects.
Next, Godson investigated lipoxin A4’s effects in animal models. Mice fed a high-fat diet become obese and develop kidney and liver disease similar to that seen in obese humans. Treating the animals with lipoxin A4 protects their livers and kidneys. Godson and her colleagues have also found that lipoxin A4 can reverse atherosclerosis in the overfed mice.
To translate those findings into practical medications, Godson is now working with synthetic chemists to develop drugs that can mimic the effects of lipoxin A4 in humans.The team has developed several promising leads, and is now testing them in various preclinical models.
Targeting the Endocannabinoid-Specialized Pro-resolution Mediator Pathway with Lenabasum to Treat Chronic Inflammatory/Fibrotic Diseases
Mark Tepper from Corbus Pharmaceuticals discussed lenabasum, the company’s synthetic, oral, small-molecule, selective cannabinoid receptor type 2 (CB2) drug. The endocannabinoid system activates the resolution of inflammation through the CB2 receptor. This GPCR is commonly found on activated immune cells during inflammation.
Based on promising preclinical results, Corbus tested lenabasum in the human clinical study (“blister model”) that has been designed to follow the progression of an inflammatory stimulus and subsequent resolution of inflammation. This study showed that lenabasum indeed stimulates inflammation resolution, reduces pro-inflammatory mediators, and promotes the clearance of bacterial endotoxins.
Corbus is now conducting more advanced clinical trials in patients with various chronic inflammatory conditions, including scleroderma, dermatomyositis, cystic fibrosis, and lupus. So far, the safety profile has been favorable with no serious adverse events attributed to lenabasum and no evidence of immunosuppression.
The Role of Resolution Phase Mediators in Oral Medicine
Thomas Van Dyke, from the Forsyth Center for Clinical and Translational Research, gave the final presentation: a look inside the mouth. Over 90% of American adults over age 30 suffer from gingivitis, and about 14% of them – or 22 million people – have severe periodontal disease. Conventional anti-inflammatory drugs can slow the progression of this disease, but can’t stop it.
A resolvin-containing mouthwash treats periodontal disease in an animal model.
In a rabbit model of periodontitis, Van Dyke has found that rinsing the animals’ mouths with a SPM analog called benzo-lipoxin A4 reverses periodontitis and restores lost bone in the jaw. That’s far better than any previously studied treatment. “There is no [prior] example of pharmacologically induced bone regeneration in periodontal disease anywhere, this just doesn’t happen,” said Van Dyke. Additional studies in pigs yielded similar results, and Van Dyke and his colleagues are now conducting a clinical trial on benzo-lipoxin A4-containing mouthwash in patients with periodontitis.
Panel Discussion: Barriers to Translation in Inflammatory Disease
The meeting concluded with a wide-ranging panel discussion with extensive audience interaction, featuring speakers Godson, Sime, Conte, Tepper, and Van Dyke, and led by keynote presenter Serhan. Panelists covered issues that ranged from the need for better biomarkers to follow inflammation resolution in laboratory and clinical studies; to the difficulty of overcoming old paradigms of anti-inflammatory interventions in medicine; and the complex interplay between drug-based and “alternative medicine” strategies for promoting inflammation resolution.
Alzheimer’s disease (AD) is a neurodegenerative disorder most common among older adults. Bruce T. Lamb, PhD, is a researcher working to unravel the causes of AD.
Published August 22, 2018
By Andre Legaspi
What inspired you to research AD?
Initially, I worked at a lab focused on Down syndrome. One of the characteristics associated with the condition is an increased risk for AD. I was fascinated as to the why, so my career started trying to understand genes, like the amyloid precursor protein gene.
Since then, I’ve become more interested in the impact that AD has on society and how prevalent the disease will be in the coming decades. As part of this, I have been a strong advocate for increased funding for Alzheimer’s disease research, including organizing a cross-country bicycle ride called the Alzheimer’s Breakthrough Ride that helped lead to the passage of the National Alzheimer’s Project Act. Much more recently, it’s become personal. I have an in-law and parent, who have mild cognitive issues related to AD.
Why are your recent findings regarding the protein TREM2* so significant?
One of the important discoveries we’ve published, about TREM2 focused therapies, is that TREM2 has both age-related and pathology-related roles in disease mechanisms. For example, we found that TREM2 plays a detrimental role early and a protective role later in terms of development of Alzheimer’s disease pathologies. That’s important because it suggests that TREM2 may have a protective role at one stage and a detrimental role at another stage later, which relates to an acute versus chronic condition.
If we want to develop TREM2-focused therapies, we need to understand what stage of AD we are targeting and how to target it. This means that we will need to carefully identify the disease stage of AD patients for immune-focused therapies.
*TREM2 is found on the brain’s immune cells.
What is the MODEL-AD consortium and how does it help further the development of AD treatments?
Bruce T. Lamb, PhD
The major goal of the consortium is to develop animal models. A 2015 NIH sponsored research meeting on AD identified the lack of accurate animal models as one roadblock in the development of potential treatments. Animal models allow us to study the development and progression of diseases and to test new treatments before they are given to people. It was appreciated that while we had a lot of decent models for early-onset AD, we didn’t have a lot of good models to decipher the more common late-onset AD.
During the initial five year funding period we hope to generate up to 50 new models and release the animals and data to the wider community as quickly as possible. The goal is to make the models available for use in understanding AD mechanisms and for testing candidate therapeutics.
Is it more realistic for a researcher to look into disease modifying therapies rather than a cure?
We’d love to find a cure. But if you look at the demographics of AD, researchers could make a huge impact by delaying its onset. If you delay the onset by five years, incidence would be reduced by half, which would have huge implications for public health. My research is concerned with trying to understand AD mechanisms and then develop therapies targeting these mechanisms.
While there is still some hope for anti-amyloid therapies and anti-tau therapies, we need to identify the next target while clinical trials for those therapies move forward. This is where immune pathways come in as there’s abundant evidence from genetics and systems biology that these are promising pathways for late onset AD.
What will the next decade bring in the treatment of AD?
We’re headed to more precision medicine type approaches. That means we will identify patients based on either genetics or bio-markers to identify a unique population and deliver therapies specified for their biomarkers. This is already happening with some of the anti-amyloid therapies, and patients are being selected based on amyloid PET scans, or CSF measures.
Approximately 36.7 million people currently live with HIV worldwide. Successful treatment with antiretroviral therapy has controlled the virus and prevented transmission in more than 20 million people. However, barriers to treatment and prevention such as stigma and discrimination, especially against women, are still prevalent in the communities most affected by HIV. According to the United Nations, an integrative strategy is required, not only in response to HIV, but also for the advancement of women’s sexual and reproductive health and rights. To accomplish this, they have focused on three main areas of intersection between HIV and women’s health: hormonal contraception, cervical cancer, and female genital schistosomiasis (FGS). Recent scientific advances raise the possibility of enhancing women’s health through closer collaboration and engagement between women, their health care providers and health programmers, and policy makers.
On March 15, 2018, the Joint United Nations Programme on HIV/AIDS (UNAIDS), the World Health Organization (WHO), and the Microbiology & Infectious Diseases Discussion Group at the New York Academy of Sciences presented Improving Women’s Health: HIV, Contraception, Cervical Cancer and Schistosomiasis. Coinciding with the 62nd session of the UN, the daylong symposium focused on improving women’s health in line with the UN Sustainable Development Goals designed to broaden community response to HIV and identify methods of intervention and treatment for reproductive diseases that afflict women.
Speakers
Sharon Achilles, MD, PhD University of Pittsburgh
Shona Dalal, PhD World Health Organization
Jennifer Downs, MD, PHD Weill Cornell Medical College
Danielle Engel, MA United Nations Population Fund (UNFPA)
Mary Rose Giattas, MD Jhpiego
Ebony Johnson, MHS Global Coalition on Women and AIDS, Athena Network
Eyrun Kjetland, MD, PhD University of KwaZulu-Natal, South Africa
Erna Milunka Kojic, MD Mount Sinai St. Luke’s, Mount Sinai West
Terry McGovern, JD Columbia University Mailman School of Public Health
Pragna Patel, MD US Centers for Disease Control and Prevention
Chelsea Polis, PhD Guttmacher Institute
Nelly Rwamba Mugo, MD Kenya Medical Research Institute
Vikrant Sahasrabuddhe, MBBS, MPH, DrPH National Cancer Institute, NIH
Annah Sango Zimbabwe Young Positives
Sponsors
Additional support provided by a medical education grant from Gilead
Session 1: Setting the Scene
Speakers
Terry McGovern, JD Columbia University Mailman School of Public Health
Women’s Sexual and Reproductive Health and the Sustainable Development Goals (SDGs)
Terry McGovern, of the Mailman School of Public Health at Columbia University, introduced the Sustainable Development Goals (SDGs) set by the UN and aimed at ending poverty by tackling a range of social and economic issues, including gender inequality. As a poverty lawyer in the late 1980s, McGovern witnessed first-hand human rights violations against women with HIV, including refusal to provide health benefits. “Science is affected by structural barriers,” said McGovern. At the time, “science had overlooked converging epidemics.” Today, the intersection between HIV and women’s healthcare, including hormonal contraception, cervical cancer, and female genital schistosomiasis continues to pose a problem in countries around the world. The purpose of this meeting was to raise awareness and promote efforts to end gender inequalities, empower women and girls, and to highlight the co-infections of HIV and female reproductive illnesses.
Speaker Presentation
Session 2: HIV and Cervical Cancer
Speakers
Danielle Engel, MA United Nations Population Fund (UNFPA)
Mary Rose Giattas, MD Jhpiego
Ebony Johnson, MHS Global Coalition on Women and AIDS, Athena Network
Erna Milunka Kojic, MD Mount Sinai St. Luke’s, Mount Sinai West
Vikrant Sahasrabuddhe, MBBS, MPH, DrPH National Cancer Institute, NIH
Annah Sango Zimbabwe Young Positives
The Intersection of Cervical Cancer and HIV
Vikrant Sahasrabuddhe, of the National Cancer Institute at the NIH, explained the intersection of cervical cancer and HIV. Cervical cancer, caused by oncogenic genotypes of the human papillomavirus (HPV), is a leading cause of cancer-related morbidity and mortality among women globally. HIV-infected women have three to five times higher risk for HPV-induced cervical lesions to progress to invasive cervical cancer. Millions of women with HIV now access affordable antiretroviral therapy globally and are living long enough for cervical cancer to manifest and progress. There are several approaches to prevent cervical cancer, including HPV vaccination and early detection and treatment of cervical precancerous lesions. Sahasrabuddhe talked about efforts underway to innovatively expand cervical cancer prevention services globally piggybacking on infrastructures developed through HIV care and treatment programs. ‘Screen-and-treat’ strategies use a low-cost simple clinical innovation called visual inspection with acetic acid (VIA) to detect precancerous lesions, followed by either freezing of the lesions by cryotherapy or referral of women with larger lesions for further evaluation and excisional treatment. Rapid, point-of-care HPV tests with self-collection of samples are being introduced for improving the sensitivity of VIA-based screening, although testing costs are still a barrier. Sahasrabuddhe concluded by emphasizing the need for improving current interventions and provided an overview of efforts for evaluating reduced dosing of HPV vaccines, improvements in screening accuracy via technological advancements, and novel non-surgical alternatives for treating precancer.
The three pillars of cervical cancer prevention provided by the WHO.
HPV Vaccine and HIV in Women
Erna Mulinka Kojic, of Mount Sinai St. Luke’s, continued the conversation on HIV and HPV co-infections. She pointed out that cervical cancer is not the only known cancer caused by HPV. For example, HPV also causes oropharynx, anal, oral cavity, larynx, vulva, and penile cancers but they have been neglected in the field, resulting in a lack of national recommendations for screening methods. Kojic then shifted the focus to HPV vaccines, which consist of virus-like particles housed in empty shells that are specific to HPV types. This specificity makes it difficult to create one vaccine for treating all nine types of HPV. Efficacy trials of the current quadrivalent HPV vaccine –which targets HPV types 6, 11, 16, and 18—indicate that it is 98% effective in preventing cervical cancer. However, in individuals previously infected with HPV types 16 or 18, the efficacy drops to 44% and even further to 17% in those with a history of any other type of HPV. Hence, the goal is to vaccinate boys and girls between the ages of 9-26 to catch cases before infection occurs. In a study on the immunogenicity of the quadrivalent HPV vaccine in HIV+ women, Kojic and colleagues found the vaccine to be highly safe and effective among this population. Vaccinated women showed a large increase of antibody titers compared to pre-inoculation levels, indicating the success of the vaccine. These exciting results present the possibility that the vaccine can do more than prevent HPV in uninfected individuals but also protect HIV+ women.
Bending the Curve for Cervical Cancer Prevention in Tanzania-Reflections on Effective Programming for Population Coverage
Mary Rose Giattas, of Jhpiego (a Johns Hopkins University affiliate), reflected on the challenges Tanzania faced while developing a national program for cervical cancer prevention (CECAP). The combination of Tanzania’s high incidence rate of cervical cancer, a large pool of adult women with HIV infection, and the lack of CECAP services provided in rural areas where more than 71% of the population lives, led the Tanzanian ministry to improve cancer services throughout the nation. In 2008, the Reproductive Health Cancer Unit was established with the goal to ensure women are free of cervical cancer. Through the support of the WHO and Merck, in collaboration with cervical cancer program implementing partners Tanzania developed a strategic plan focusing on the WHO’s three pillars of prevention. Jhpiego with USG funded MAISHA program provided technical inputs in the development process. While this plan has allowed doctors to successfully treat patients with small lesions using cryotherapy, the limited supply of LEEP devices creates a challenge in treating patients with larger lesions. Hence, collaboration between the Tanzanian ministry and Jhpiego, an international non-profit health organization with USG funding, has helped to further strengthen the key health system components in the program with the focus on the same day screening and treatment . Jhpiego has also been providing technical assistance to other implementing partners to improve access and quality of cervical cancer prevention services including timely and appropriate referral. Strong partnership between the MOH and implementing partners has contributed to scale up high quality VIA and Cryotherapy services for women with cervical cancer. In the 9 years since it was first implemented, this program has increased the number of CECAP sites from 3 to 557, improved cervical cancer and HIV testing, and reached more than 600,000 women in Tanzania. Bending the curve for cervical cancer prevention in Tanzania requires improving coverage by providing greater access to services through health system strengthening led by an empowered MOH, which has taken on the task of eliminating cervical cancer in Tanzania.
Cervical and uterine cancer affect approximately 38% of women in Tanzania.
How Does Science and Delivery Affect People in Communities?
Following the incredible milestones of Tanzania’s health services, Annah Sango, of the Zimbabwe Young Positives, spoke about the harsh realities women in Zimbabwe face when seeking medical treatment for HIV. Some common issues include long lines at doctors’ offices due to limited supplies, low accessibility of healthcare providers in rural areas, and the stigma prevalent throughout the community. She advocated for more information on medical treatments, more access to care in both urban and rural areas, the need to involve and educate young boys and men, and friendlier doctors who provide unbiased treatment. Government collaborations with health organizations, similar to the programs Tanzania successfully implemented, would greatly benefit the Zimbabwean community.
The United Nations Joint Global Programme on Cervical Cancer
Danielle Engel, of the United Nations Population Fund, presented the development of a new, five-year Joint Programme at the UN, which seeks to eliminate cervical cancer globally. Given that we know how to treat cervical cancer and the prevention is cost effective, the greatest obstacle is making treatment widely available, especially in middle- and low-income nations with the highest incidence rates. The Programme is based on three pillars: prevention, treatment, and care. At the national level, the goals are to increase HPV immunization access to all girls, ensure screening and treatment for cervical pre-cancer by providing technical assistance to local governments, and guarantee universal access for the diagnosis and treatment of invasive cervical cancer. At the global level, new innovative technologies are needed for cervical cancer screening, as well as policies to improve and increase access to HPV vaccination.
The burden of cervical cancer is highest in the global south, with sub-Saharan Africa particularly affected.
Women, HIV, HPV and Cervical Cancer: An Intersectional Approach Moving beyond the Biomedical Lens
Although technological advances have enabled a needed reduction of new HIV infections, these advances are hampered by policy, funding, and access barriers in many parts of the world. Ebony Johnson, from the Global Coalition on Women and AIDS, Athena Network, gave a compelling call to action, emphasizing the need to go beyond the biomedical to include a human rights lens in the creation of enabling environments for healthy women and girls. As part of the Athena Network’s global #WhatWomenWant campaign, Johnson and colleagues asked women and girls around the world what they need to be safe, healthy, educated, and empowered in their communities. Participants said they want respect, representation, equal opportunities, power and autonomy over their own bodies, and much more. Johnson advocated for women and girls to be a part of research at the onset and for doctors to communicate better with their patients, creating bi-directional interactions and suspending judgment. “[Women] know our bodies, we know our communities, we know our environments, we know our barriers,” said Johnson, “and we know how to help you help us.” Ultimately, expanding funding, igniting political will, promoting intersectional sexual and reproductive healthcare, and developing innovative HIV, HPV and cervical cancer programming are key components of achieving gender equality.
Session 3: The Right to Comprehensive Sexual and Reproductive Health and Rights
Speakers
Peter Godfrey-Faussett UNAIDS
Sharon Achilles, MD, PhD University of Pittsburgh
Shona Dalal, PhD World Health Organization
Ebony Johnson, MHS Global Coalition on Women and AIDS, Athena Network
Nelly Rwamba Mugo, MD Kenya Medical Research Institute
Chelsea Polis, PhD Guttmacher Institute
Vikrant Sahasrabuddhe, MBBS, MPH, DrPH National Cancer Institute, NIH
Annah Sango Zimbabwe Young Positives
Hormonal Contraceptive Methods and Women’s Risk of HIV Acquisition: Understanding the Evidence
Observational studies have suggested an association between use of specific hormonal contraceptives (HCs), particularly the injectable depot medroxyprogesterone acetate (DMPA), and increased risk of HIV acquisition in women. This issue is critically important for women’s health, particularly in sub-Saharan Africa, where high rates of HIV coincide with high use of injectable contraception. Chelsea Polis, from the Guttmacher Institute, presented the results of a comprehensive review she conducted with her colleagues on the relationship between HC methods and HIV risk. The preponderance of data for oral contraceptive pills, injectable norethisterone enanthate, and levonorgestrel implants do not suggest an association with HIV acquisition, though data for some of these methods were limited. On the other hand, new information increases concerns about a potential causal association between DMPA and HIV acquisition risk in women, although the possibility of confounding in these observational data cannot be excluded. Since DMPA is the most commonly used type of birth control in sub-Saharan Africa, these results could have important implications for these epidemiological populations. A meta-analysis estimated that DMPA may increase a woman’s risk of HIV acquisition by about 40%. In other words, if the association between DMPA and HIV is causal, a woman with a 2.4% chance of HIV per year would increase her risk to 3.3% if she uses DMPA. Similarly, a woman who starts at a higher baseline risk, for example, 14% per year, would increase her risk to about 19% per year if she uses DMPA. While these studies suggest a potential causal association between DMPA and women’s risk of HIV, all currently available data are observational, and thus potentially vulnerable to confounding. A randomized study is underway to attempt to parse out the relationship between various HC methods and women’s risk of HIV acquisition.
Even though Sub-Saharan Africa has the lowest contraceptive use compared to other regions, injectables comprise the largest portion of contraceptive methods. The prevalence of injectables is due to their discrete and easy-to-use nature, but it is concerning that their use overlaps with nations where HIV prevalence is highest.
Hormonal Contraception and HIV Acquisition Risk: Biological Plausibility, Clinical Evidence, and Research Gaps
Sharon Achilles, of the University of Pittsburgh, addressed the biological evidence for the possible mechanisms of action of HCs in HIV-infected individuals. She focused on four main mechanisms, including: changes in architectural epithelial cells, target cells, immune changes, and microbiota. Architectural changes of epithelial cell thinning were seen in animals given DMPA. However, similar studies in women failed to show significant thinning between the follicular and luteal cycles, which has led researchers to nearly abandon this mechanism of action. Evidence for target cell changes in healthy HIV- women on DMPA showed increased cervical CD8CCR5+ target cells during high progestin concentrations, which was not evident in women using Net-En, an alternative injectable contraceptive. Studies on adaptive and innate immune changes have provided variable results making it difficult to converge on a specific mechanism. Finally, none of the hormonal methods shifted the vaginal microbiota studied including Lactobacilli, Gardnerella, and Atropobiumvaginalis. Achilles cautioned that the study of biological changes associated with HC is complicated by the diversity of available hormones and delivery routes. There is a need for more precise categorization of progestin types, progestin concentration, and mode of administration to converge results from multiple studies and understand the biological mechanisms linking HCs to HIV.
Update on the Evidence for Contraceptive Options and HIV Outcomes (ECHO) Trial
Given the lack of a causal link between DMPA and HIV risk, the Evidence for Contraceptive Options and HIV Outcomes (ECHO) trial was created to fill this critical knowledge gap. Nelly Rwamba Mugo, of the Kenya Medical Research Institute, provided an update and overview of the trial. As an open-label randomized clinical trial for HCs, ECHO has recruited more than 7,800 women, ages 16-35, in four countries since it began in 2015. Participants were randomly assigned to one of three types of birth control: DMPA, levonorgestrel (LNG) implant, or copper intrauterine device (IUD), and regularly tested over 18 months for HIV and pregnancy. Furthermore, participants were given contraceptive and HIV-risk reduction counseling. The results of the study, expected within the next year, will indicate HIV incidence, pregnancy rates, serious adverse events (SAEs), and method continuation associated with each of the three HCs. This information will help policymakers formulate counseling options for clinicians and educate women and communities on the benefits and risks of using the three contraceptive methods.
The ECHO trial will allow researchers to compare HIV incidence across three different types of contraceptives using twelve study sites across four African nations.
Translating Evidence to Policy: Guidance on Hormonal Contraceptive Use for Women at High Risk of HIV
Shona Dalal, of the World Health Organization (WHO), reviewed the Medical Eligibility Criteria (MEC) used by the WHO to provide recommendations on 25 methods of contraception. Based on the review of various hormonal contraceptive methods and HIV conducted by Chelsea Polis and colleagues, the WHO changed the classification for progestogen-only contraceptives for women at high risk for HIV from no restriction, to an indication that the advantages of this method generally outweigh the theoretical risks. In reassessing the risk level, they considered the following: quality of the evidence (GRADE profile), values and preferences of contraceptive users and providers; balance of benefits and harms; priority of the problem; equity and human rights; acceptability; and feasibility. Although there continues to be evidence of a possible increased risk of contracting HIV among progestogen-only injectable users, there is an absence of evidence from randomized trials and unclear causality (methodological issues versus a real biological effect). Dalal stressed that messaging surrounding possible risk is critical so that women at low risk of HIV infection don’t change their preferred method of contraception. “The WHO is committed to keeping emerging evidence under close review,” said Dalal, before explaining that data from the ECHO trial is highly anticipated so that the WHO can incorporate it into future guidance accordingly.
Panel Discussion: Integrating Rights, Services and HIV
The first panel discussion addressed the impending results of the ECHO trial and its impact on the communities that use DMPA. Peter Godfrey-Faussett from UNAIDS moderated the conversation, which included four of the speakers: Annah Sango, Ebony Johnson, Nelly Rwamba Mugo, and Vikrant Sahasrabuddhe. The panelists warned that African communities rely heavily on DMPA for contraception and it is vital that they are informed early about the possible outcomes of the study and how to proceed. Education about alternative methods of contraception should come from a collaborative partnership between patients and healthcare providers rather than media outlets. This can prevent fear and overgeneralizations of HIV risk to other types of birth control which may not apply.
Panelists also discussed the intersection of HPV, contraceptives, and cervical cancer. Sahasrabuddhe noted that the literature on HCs and HPV acquisition is mixed but oral contraceptives tend to increase risk of HPV. Yet the studies conducted on these co-factors are mostly observational and should be revisited. On the other hand, the known progression of HPV to cervical cancer provides opportunities for developing better screening techniques and possibly a single dose, multivalent vaccine that would protect against 98% of HPV types. The session closed with take-home points emphasizing the need for better doctor-patient interactions, good policies translating to good practice, and keeping the public scientifically educated.
Am J Obstet Gynecol. 2018 Mar 2. pii: S0002-9378(18)30176-5.
Session 4: Female Genital Schistosomiasis (FGS)
Speakers
Jennifer Downs, MD, PHD Weill Cornell Medical College
Eyrun Kjetland, MD, PhD University of KwaZulu-Natal, South Africa
Pragna Patel, MD US Centers for Disease Control and Prevention
Female Genital Schistosomiasis: A Burning Sensation, Malodorous Discharge and Bleeding from a Waterborne Parasite
The final session of the symposium shifted towards the intersection of HIV and Female Genital Schistosomiasis (FGS). Eyrun Kjetland, from the University of KwaZulu-Natal, South Africa, explained that urogenital schistosomiasis, a largely African, waterborne, parasitic disease that can cause Female Genital Schistosomiasis (FGS), affects the cervix, fallopian tubes, and the urinary tract. The cycle of FGS begins with eggs released with urine or feces in fresh water rivers or lakes. These eggs hatch, releasing larvae that infect snails and multiply into many cercariae parasites. The parasites then penetrate human skin and lay eggs inside the body, which typically penetrate the cervix and appear as grainy, sandy patches on the vaginal wall. In a cross-sectional study in a Zimbabwean community, 41% of women with FGS were also HIV+. Importantly, FGS, which may mimic the symptoms of cancer and sexually transmitted infections is not limited to adult women. Young girls aged 10-12 typically contract it and can develop symptoms including: intravaginal lesions, malodorous discharge, bloody discharge, a burning sensation in the genitals, and ulcers. Affected young girls grow into adulthood with damaged genitals and may experience infertility and ectopic pregnancy. Starting treatment at a young age can help reduce contact bleeding and decrease CCR5-HIV receptor expression in CD4+ cells, a receptor used by HIV to enter and infect host cells.
HIV and Schistosomiasis: Summary of the Evidence
Jennifer Downs, of Weill Cornell Medical College, summarized evidence from epidemiological studies in sub-Saharan Africa on the relationship between FGS and HIV. To date, three studies have found an increased odds of being HIV-infected of at least 2.9 in women with FGS by measuring either parasite eggs or circulating anodic antigen, a carbohydrate produced in the gut of adult schistosome worms. A similar study in men failed to find a significantly increased odds of HIV infection, highlighting that the intersection between schistosomiasis and HIV specifically affects women. Work done in macaque monkeys with pre-existing Schistosoma mansoni infections showed that when they were rectally inoculated with increasing doses of simian HIV (sHIV), they became sHIV-infected at a dose 17 times lower than macaques without schistosomiasis. Furthermore, the monkeys infected with Schistosoma mansoni showed increased HIV viral replication. Downs reported results from a nested case-control study situated within a longitudinal study in adults of reproductive age since 1994 in Kisesa, Tanzania. In the longitudinal study, participants are tested for HIV every three years and have blood spots stored for additional analyses, allowing researchers to compare individuals who recently contracted HIV to matched controls that didn’t contract HIV during this period. Downs and colleagues documented that the prevalence of the schistosome infection at the time of HIV-seroconversion was higher among HIV-1 female seroconverters but not males. Seroconverters with schistosome infection also have higher HIV-1 RNA levels at the time of HIV diagnosis. Hence, this study has been vital in extending findings on schistosomiasis from animal models to humans.
The results of a longitudinal study on schistosomiasis and HIV underscore the importance of understanding the intersection between these diseases.
Schistosomiasis and HIV Transmission: A Call for Research
The dearth of research on schistosomiasis and HIV transmission is a problem for this largely African epidemic. Schistosomiasis affects more than 230 million people worldwide, 90% of whom are in Africa. Pragna Patel, of the CDC, highlighted that there is not enough epidemiological evidence in terms of incidence and prevalence of FGS and HIV. Furthermore, due to decreasing child mortality, the youth population in sub-Saharan Africa will double by 2020 from the start of the HIV epidemic in 1990. This increase in the size of vulnerable populations and the fact that young girls typically contract FGS through freshwater contact with recurrent exposures throughout their lifespan, requires improved treatment of FGS to reduce HIV infections in the future. Patel made a call for research on the effect of FGS and its treatment on HIV acquisition, particularly among adolescent girls and young women.
Adolescent girls are a vulnerable population for HIV infection.
Next Steps: Research, Policy, Activism
The final session ended with an open discussion in which audience members dove deeper into the current understanding of schistosomiasis by asking questions on its transmission and overlap with HIV. The speakers noted that while men are less likely to have schistosomiasis, those who do, tend to have bloody semen, making it more likely that they transmit HIV. This intersection with HIV can be leveraged to provide more widespread treatment of schistosomiasis. Kjetland mentioned a current randomized trial she is conducting in African communities which uses a thorough questionnaire to identify potential confounds in kids with schistosomiasis. Such trials can catch cases at an early stage and provide consistent treatment. Downs called attention to the WHO guidelines for treating schistosomiasis, which indicate that in communities with prevalence above 50%, treatment should be provided twice a year to all community children. Yet, less is done to diagnose and treat adults with schistosomiasis. Pragna mentioned that the DREAMS initiative for adolescent girls and young women, which is currently implemented in a number of African communities, could be anchored to create an integrative package to educate women on HPV, cervical cancer, HIV, and schistosomiasis. These types of initiatives along with good policies can greatly reduce prevalence and stigma of female-reproductive diseases around the world.
Scientists, ethicists, and other experts gather to discuss the promises and potential consequences of advances in biotechnology and artificial intelligence aimed at improving human performance.
New York, NY | May 10, 2018 — From eyeglasses that restore sight to robotic prosthetics to replace limbs, people throughout history have sought to overcome the limitations of the human body. New advancements in such technologies and their implications will be explored at “The Enhanced Human: Risks and Opportunities,” presented by the Aspen Brain Institute, The Hastings Center, and The New York Academy of Sciences at the Academy’s headquarters on Monday, May 21 at 6:00pm.
This evening event will include short presentations and a panel discussion examining the scientific and ethical implications of existing and rapidly emerging technologies with applications for human enhancement. Special emphasis will be placed on CRISPR/Cas9 gene editing technology and artificial intelligence. Experts from multidisciplinary fields will provide historical perspective and scientific background before discussing the vast opportunities of these cutting edge technologies and delving into the complex ethical and social questions still to be addressed.
The program will begin with introductory sessions on “The History and Science of Human Enhancement” and “Present and Future Bioethical Considerations,” featuring brief talks from renowned geneticist George Church (Wyss Institute at Harvard University), biomedical ethics and policy expert Josephine Johnston (The Hastings Center), technology futurist Jamie Metzl (Atlantic Council), and artificial intelligence specialist Meredith Whittaker (AI Now Institute at NYU).
These introductory sessions will be followed by a lengthy panel discussion moderated by Mildred Z. Solomon, distinguished health care and science policy expert and president of The Hastings Center. The panel is comprised of the aforementioned speakers and Glenda Greenwald, president and founder of the Aspen Brain Institute. A speaker networking reception will close the event. For those unable to attend the event in person, the event will be available via Livestream.
This event was made possible, in part, through the support of a grant from the John Templeton Foundation. The opinions expressed are those of the presenters and do not necessarily reflect the views of the John Templeton Foundation.
About the Aspen Brain Institute
The Aspen Brain Institute convened its first meeting co-presented with The New York Academy of Sciences in 2010 focused on Neurotechnology: Building Better Brains. Since 2010, the Aspen Brain Institute has partnered with the Academy on six symposia and a social impact challenge. As a 501(c)(3) non-profit organization, the Aspen Brain Forum Foundation supports and produces scientific meetings covering topics ranging from neuroprosthetics to the developing human brain. The Foundation’s mission is to:
Organize, produce, and host an annual high-level meeting of international brain researchers, in partnership with The New York Academy of Sciences, leading to global collaborations and breakthroughs in world brain science.
Present and disseminate the most cutting-edge innovations in brain science.
Ally with large new initiatives, such as the American Brain Coalition, the American Brain Foundation, and One Mind for Research, to prevent and cure brain disorders such as Alzheimer’s, Parkinson’s, autism, and depression, within a decade.
About The Hastings Center
The Hastings Center addresses fundamental ethical and social issues in health care, life sciences research, and biomedical technologies. The Center’s goal is to promote compassionate and just health care and the wise use of emerging technologies. Through its scholars’ writing and speaking, and through the work of the many people from around the world who participate in its projects or submit articles to its two journals, The Hastings Center shapes ideas that influence key opinion leaders, including health policymakers, regulators, lawyers, legislators, and judges, as well as health care executives, physicians and nurses. Founded in 1969 by philosopher Daniel Callahan and psychoanalyst Willard Gaylin, The Hastings Center is the oldest independent, nonpartisan, interdisciplinary research institute of its kind in the world. In addition to producing original research, it accomplishes its mission through public engagement and service to the field of bioethics. To learn more, please visit www.thehastingscenter.org/.
The Innovators in Science Award Honorees are Breaking New Ground in Neuroscience: Dr. Shigetada Nakanishi has uncovered essential components of neural networks.
Published May 1, 2018
By Anni Griswold
Albert Einstein reportedly once said, “Not everything that can be counted counts, and not everything that counts can be counted.” Though the 2017 honorees of the Innovators in Science Award have plenty of countable achievements, their stories reveal a common thread — creative approaches to their work and the development of disruptive tools that transformed scientific understanding in their discipline.
Unmasking Cellular Messengers
Shigetada Nakanishi
During medical school, Shigetada Nakanishi, MD, PhD, became frustrated when he realized how little was known about the etiology of many diseases. “As a consequence, I gradually began to think that research work on basic medicine to explore the mechanisms of diseases is more valuable as my life work,” he says.
This change of heart set him on a path of scientific discovery. It eventually shaped our modern understanding of the brain’s function. Nakanishi is Director of the Suntory Foundation for Life Sciences Bioorganic Research Institute and Senior Scientist Winner. He has uncovered essential components of neural networks, including diverse glutamate receptors that mediate communication between neurons. His work has also revealed how the cerebellar and basal ganglia circuits control motor coordination, learning and motivation.
Along the way, he developed an innovative cloning strategy for cloning membrane-embedded transmitter receptors, and uncovered genes encoding NMDA and G-protein coupled glutamate receptors.
“Science can be fruitfully done and [is] enjoyable when you design and carry out your experiments according to your own questions and ideas,” he says. “Then, you will be deeply inspired and surprised with the beauty of nature.”
Read more about Innovators in Science Award Honorees:
Researchers across the globe are doing their part to both fuel and sustain a healthy planet.
Published May 1, 2018
By Hallie Kapner
Patrick Schnable
To the untrained eye, the black dots speckling the corn leaves in the greenhouses at Iowa State University’s Plant Sciences Institute could be anything — blight, mold, rot. But to Patrick Schnable, the Institute’s director and the C.F. Curtiss Distinguished Professor and Iowa Corn Endowed Chair in Genetics at ISU, the dots are the future of precision irrigation — a simple and inexpensive window into how plants use a precious global resource: water.
Dubbed the “plant tattoo,” the dots are bits of graphene oxide deposited on a gas-permeable tape to form an easily applied sensor that precisely measures transpiration — water loss — on an individual-leaf basis. As leaves lose water, the moisture changes graphene’s electrical conductivity. By measuring those changes, Schnable and his collaborators can observe transpiration in real time.
“If you have a plant under drought stress and you water it or it rains, you can track water moving up through the plant,” Schnable said. “For the first time ever, we can observe plants reacting to an irrigation event as it happens.”
The plant tattoo is one of countless research initiatives underway worldwide that aim to conserve and maximize natural resources, improve access to nutrition, prevent and treat disease, and boost the health and well-being of the planet’s people and wildlife.
Schnable and his collaborator, Liang Dong, associate professor of electrical and computer engineering at ISU, envision a day when farmers can use plant sensors to guide irrigation decisions and breeders can use them to create drought-resistant varietals. The researchers are already adapting the technology for use beyond the Iowa cornfields. While the current version requires connection to a control box to provide both voltage and transpiration rate analysis, plant tattoo 2.0 will be wireless and smartphone-compatible. Such refinements will drop the cost of the system even further, making the sensors accessible for areas of the developing world where every drop of water counts.
Cultivating “Black Rice”
Ujjawal Kr. S. Kushwaha
Maximizing efficiencies in breeding and irrigation of agricultural crops is one key part of meeting the global goals related to hunger, nutrition and stewardship of the land. Equally critical are efforts to identify and promote staple crops that pack maximum nutrition, explained Ujjawal Kr. S. Kushwaha, PhD Scholar in Genetics and Plant Breeding at G.B. Pant University of Agriculture and Technology in Pantnagar, India.
More than half of the world’s population relies on rice for at least 20 percent of their daily calories. If Kushwaha had his way, the typical white rice of subsistence would be replaced by black rice, an heirloom variety sometimes called “forbidden” rice, and one of nature’s nutritional powerhouses.
“No other rice has higher nutritional content,” Kushwaha said. “It’s high in fiber, anthocyanins and other antioxidants, vitamins B and E, iron, thiamine, magnesium, niacin and phosphorous. Consumed at scale, it could have a significant impact on malnutrition.”
Decades of effort to boost the nutritional content of rice have yielded biofortified varietals rich in iron, zinc and provitamin A. While addressing these highly prevalent micronutrient deficiencies is critical, Kushwaha contends that black rice could address both a broad spectrum of nutritional deficiencies as well as provide anti-inflammatory and anti-atherogenic benefits.
However, black rice is not widely cultivated outside of China, and most varietals are relatively low-yield, which drives the crop’s high cost. Kushwaha is working to shift that equation, spreading the black rice gospel with the hope of boosting demand and incentives for farmers to develop higher-yield varietals, which could make a crop once reserved for royalty as affordable as white rice.
Anticipating the potential hurdles of acceptance — factors such as taste and color often determine whether new varietals are adopted or rejected — Kushwaha and others cultivating nutrient-rich rices have determined that black rice could be bred to minimize color while preserving much of its nutritional value. “Some of the qualities could be reduced, but it’s still far better than white rice,” he noted.
Plant Power
Plants already do far more than just feed the world — we derive fuel, fabrics, medicinal compounds and much more from them. Yet over the past two decades, a new role for plants has emerged — one that may revolutionize one of the most important pipelines for global health: vaccine production.
Conventional vaccine manufacturing relies on primary cells — like chicken eggs — mammalian cell lines, yeast cells or bacteria. These approaches have well-known limitations, such as long production times, variable yields and risk of contamination by other human pathogens. As Kathleen Hefferon, a virologist and Fulbright Canada Research Chair of Global Food Security at the University of Guelph explained, plants are not merely viable alternative bioreactors for many types of vaccines — they are production superstars.
First-generation plant-made biopharmaceuticals were derived from transgenic crops, but public concerns about GMOs, as well as variability in the amount of vaccine protein produced per plant, drove the development of a second — and now dominant — production method. Plant virus expression vectors are used to deliver genes for producing vaccine proteins into the leaves of plants such as tobacco and potato, turning common crops into factories capable of churning out huge quantities of vaccine protein faster and more cheaply than any other method.
Plant-made vaccine proteins carry no risk of contamination with mammalian pathogens, and better still, plants can produce similar post-translational modifications to human cells, which increases biocompatibility. Hefferon believes plant-made biopharmaceuticals will grow exponentially over the next five years, due in part to increased interest in stockpiling vaccines against pandemic flu and other diseases.
“It’s hard to stockpile vaccines produced in mammalian systems, and it’s very hard to produce enough vaccine in time to be helpful in an outbreak,” she said. “Plants offer a clear advantage here.”
Several pharmaceutical companies have plant-made vaccines and therapeutics in clinical trials, but the public is already familiar with one experimental drug that made headlines in 2015 — ZMapp, which was used to treat several Ebola-infected healthcare workers in West Africa. Hefferon is also quick to emphasize that the lower-cost profile of plant-made vaccines has special relevance for cancer prevention in the developing world, where rates of cancers linked to vaccine-preventable viruses, including HPV, are skyrocketing.
“We’re already in the running to advance the science toward pharmaceutical production in plants,” she said. “The current systems have so many limitations and plants are an incredible alternative.”
On Land and Sea
Just as human health is inextricably tied to the health of the air, soil, water and environment, so too is the health of the animals we rely on for work and food. In the tropical regions of Mexico, scientists including veterinarians Felipe Torres-Acosta and Carlos Sandoval-Castro, and organic chemist Gabriela Mancilla, of Universidad Autonoma de Yucatan (UADY), are studying how sheep and goats regulate their own health through diet.
The team at UADY has been devising strategies to improve the health of ruminants in tropical environments for 30 years. One of their standout findings is that malnourished animals are less resilient to native parasites, and while farmers can boost resilience with supplemental food, access to native flora is critical for keeping the host-parasite relationship in balance.
The UADY team showed that sheep and goats left to forage on their own in the Mexican jungle feast on an astonishing 60 different plant species per day, adjusting their food choices based on seasonal availability. Diving deeper into the connection between diet and immune resistance, Torres-Acosta’s team collected samples of ruminants’ preferred foods, analyzing them for nutritional content and the presence of anthelmintic activity.
Stephen Frattinii Photo: Hudson Rivers Fisheries Unit Staff
Analysis reveals that most local flora do contain anti-parasitic compounds, and Mancilla is working to discover the mechanisms by which they act to control parasite load. The team is investigating whether animals intentionally seek a diet rich in plants that naturally limit parasite infection. This work, as well as similar research in sheep and goats around the world, is already impacting how some small farmers treat infections.
“If animals have access to their native foods, they can keep parasites in check, which reduces the need for medication and allows farmers to treat only the sickest animals,” Torres-Acosta said. “The most interesting things we’re learning come directly from observing the animals — given the choice, animals know what they need to eat to stay healthy, and we can learn so much from their innate wisdom.”
Off the shores of Long Island, New York, Stephen Frattini, founder of the Center for Aquatic Animal Research and Management (CFAARM), is trying to bring a similar sensibility to the seafood industry, which supplies three billion people worldwide with their primary source of protein. Frattini, a veterinarian, focuses not just on how fisheries and aquaculture operations could improve fish welfare, though his passion for that subject runs deep.
His goals are bigger, and include uniting experts in animal welfare, engineering, health management, feed development and consumer psychology to transform the seafood industry from a profoundly siloed one, rife with inefficiencies and transparency issues, to an integrated one that places the health of the environment, people and fish front and center. Frattini believes that a more integrated seafood industry could revitalize coastal communities both in the United States and developing countries, as well as advance production strategies already known to improve fish health, such as emphasizing diversity over monoculture.
“We still need a much better understanding of fish behavior in captivity and what we can do to create happier, healthier animals, but I’m convinced we can increase efficiencies while increasing fish contentment, which is a win for animals, the environment and the industry,” he said.
A Matter of Will
William Haseltine
Decades of fast-paced discovery in medical research, coupled with high-tech advances in equipment, procedures and information technologies have yielded many of the solutions necessary to provide high-quality healthcare to all. No cohort in history has been better equipped than ours to identify problems, connect patients with preventative and acute care and measure and understand the outcomes. Yet nations around the globe, from the most developed to the least, struggle to manage the cost, logistics and delivery of basic human health services.
A desire to identify best practices and help spread their adoption drove William Haseltine, a biologist and former professor at Harvard Medical School, known for his pioneering research on HIV/AIDS and the human genome, to found the nonprofit ACCESS Health International 10 years ago.
ACCESS Health has since partnered with nations in every region of the world to better understand the systems that improve primary care, lower maternal and child mortality, and meet the needs of an aging population while maintaining affordability. From a revolutionary emergency-response system in India that serves 700 million people each year with greater efficiency and lower cost than any system in the West, to hospitals using information technology to implement radical transparency and accountability systems that are improving patient safety, Haseltine and the ACCESS Health team have found no shortage of strategies that save and improve lives within budget. Bringing them to bear on the global problem of healthcare access is mainly a matter of will.
“We have a lot of knowledge that can be deployed broadly across the globe, but there has to be a desire and incentive to change,” Haseltine said.
The 17 SDGs can be viewed as a tally of ways people and planet can suffer and struggle. But they can also be viewed as vision of hope, a commitment by 193 nations to alleviate pain and work toward a healthier, more equal future.
“We have come to the point where we have the ability to dramatically improve health outcomes, whether it’s in environmental health, or improving maternal and infant mortality,” said Haseltine. “It all comes down to the question: do we have the will to do it? When the answer is yes, it’s transformative.“
Drone Delivery Takes Off In Rwanda
Delivering goods via drones is not a new idea, but it’s providing an important sustainable lifeline to rural communities in Rwanda that are benefiting from the technology.
California-based automated logistics company, Zipline and the Government of Rwanda have collaborated on the world’s first national drone delivery service for on-demand emergency blood deliveries to transfusion clinics across the country. Since its launch in October, 2016, Zipline has flown more than 7,500 flights covering 300,000 km, and delivered 7,000 units of blood to physicians and medical workers in Rwandan villagers nationwide.
Zipline’s technology was developed for longer-haul flights than typical drones and have a round trip range of 160 kilometers. The drones can carry 1.5 kilos of cargo and cruise at 110 kilometers an hour.
More importantly the craft are built to handle the challenges of Rwanda’s mountainous terrain and extreme weather conditions. They look more like fixed wing airplanes than the typical quadcopter image, but it is one of the reasons why they are capable of flying faster and farther than standard craft; imperative for speeding-up the delivery of life-saving medical supplies to remote communities.
The airplanes are powered by lithium-ion battery packs. Two twin electric motors provide reliability at a low operating cost. Redundant motors, batteries, GPS and other electronics provide the safety features, in addition to a parachute-enabled landing system. The planes fly on predetermined routes and are monitored by a Zipline operator.
The Innovators in Science Award Honorees are Breaking New Ground in Neuroscience: Dr. Kay Tye has made discoveries between neural networks and social interaction.
Published May 1, 2018
By Anni Griswold
Albert Einstein reportedly once said, “Not everything that can be counted counts, and not everything that counts can be counted.” Though the 2017 honorees of the Innovators in Science Award have plenty of countable achievements, their stories reveal a common thread — creative approaches to their work and the development of disruptive tools that transformed scientific understanding in their discipline.
Bridging Psychology and Neuroscience
As an undergraduate at the Massachusetts Institute of Technology, Kay Tye, PhD, an Early-Career Scientist Finalist, enjoyed taking psychology classes alongside her load of neuroscience coursework. But the contrast revealed each field’s shortcomings. Psychology felt unsatisfying, she says, because it lacked a mechanism to trace thought and emotion back to neural mechanisms. And neuroscience focused on sensory or motor systems without hinting at how these systems give way to thought and emotion.
Eventually, she devised a plan to bridge the fields. She began using optogenetics to tease apart the underpinnings of motivation and reward. “The dream has always been to completely understand on every level how complex social and emotional representations exist in the brain,” says Tye, Assistant Professor at MIT’s Picower Institute for Learning and Memory. Using this approach, Tye has made startling discoveries about the neural networks involved in social interaction, including the finding that loneliness drives social interaction.
Going forward, she aims to explore how social representations are parsed in the brain. This research program, she says, could someday lead to targeted therapeutics for psychiatric conditions that have minimal side effects.
“If we understand the cells and circuits and synapses that give rise to different emotional states,” she says, “then we can understand when there are perturbations and how to fix them.”
Read more about Innovators in Science Award Honorees:

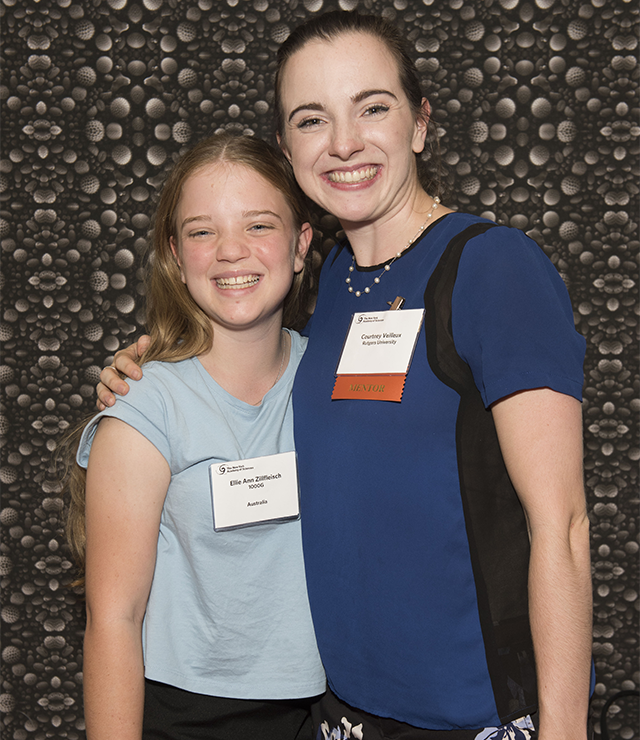





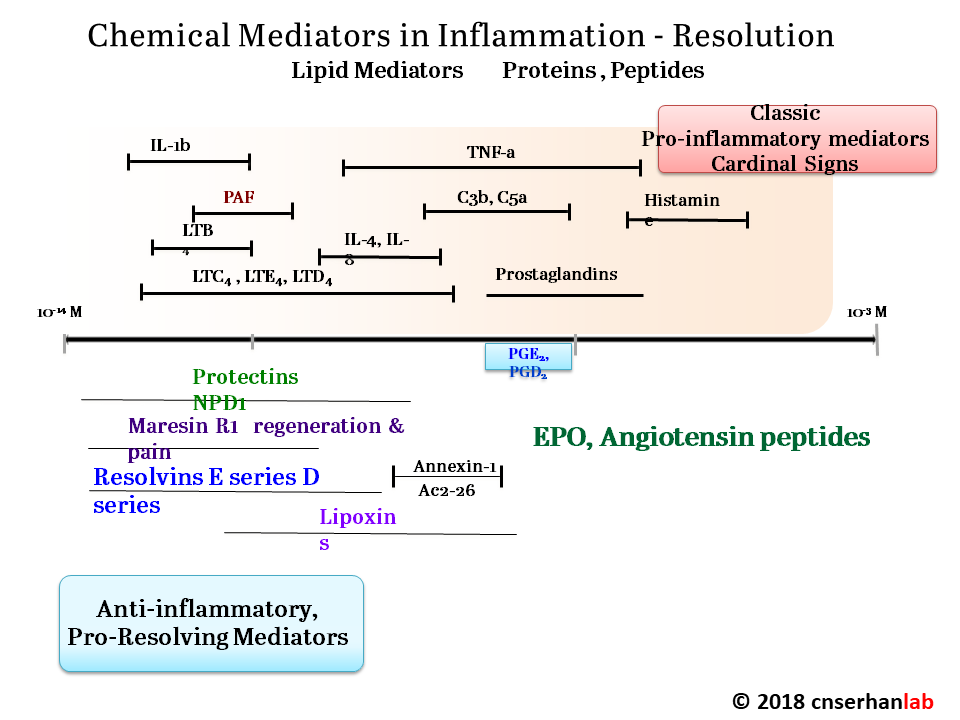
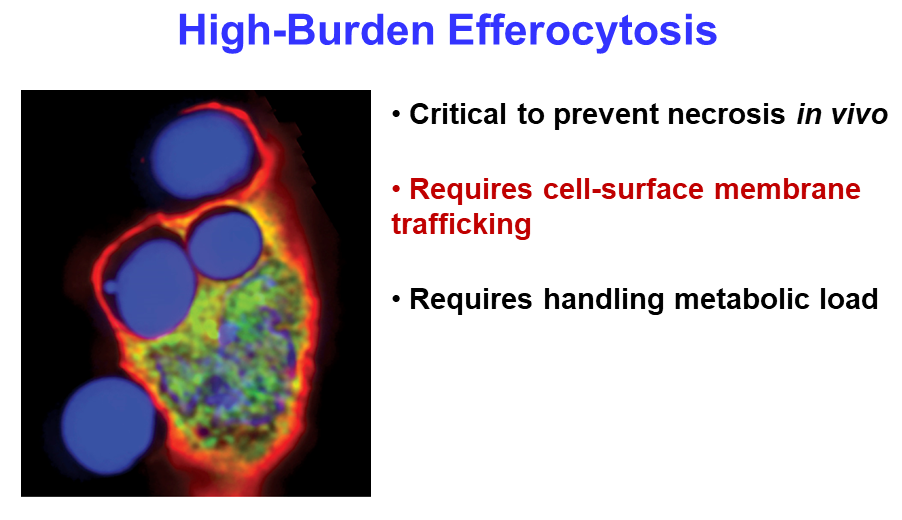
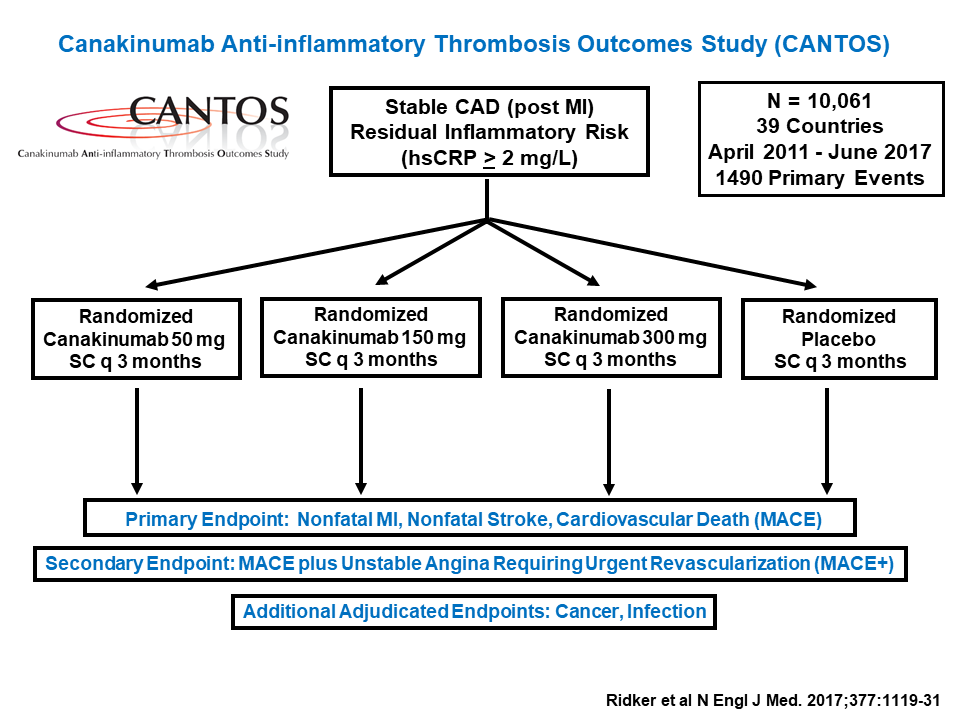

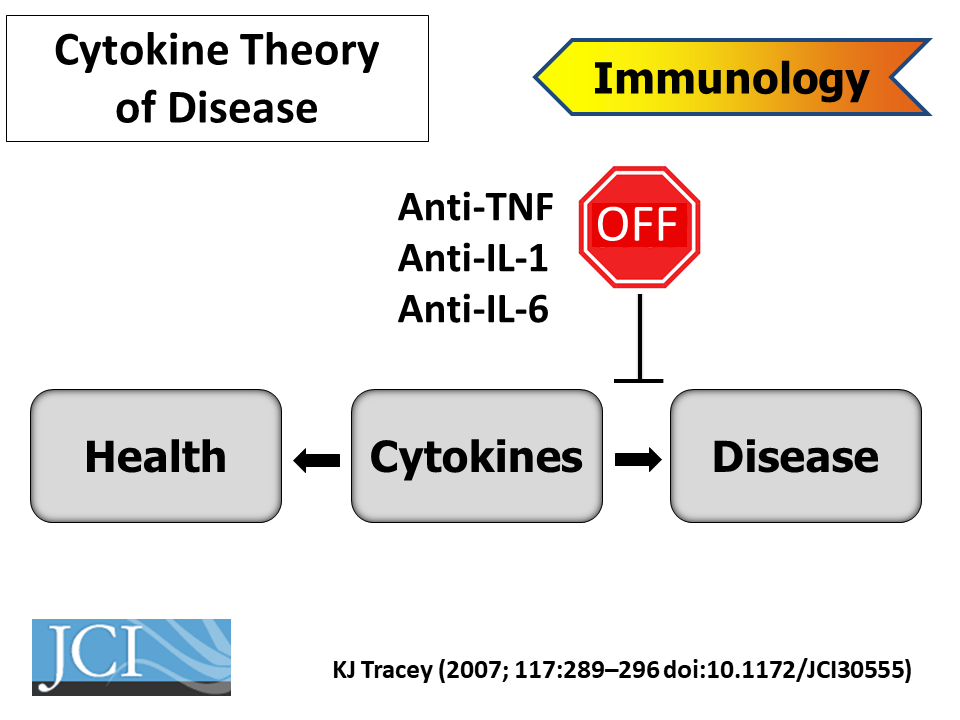

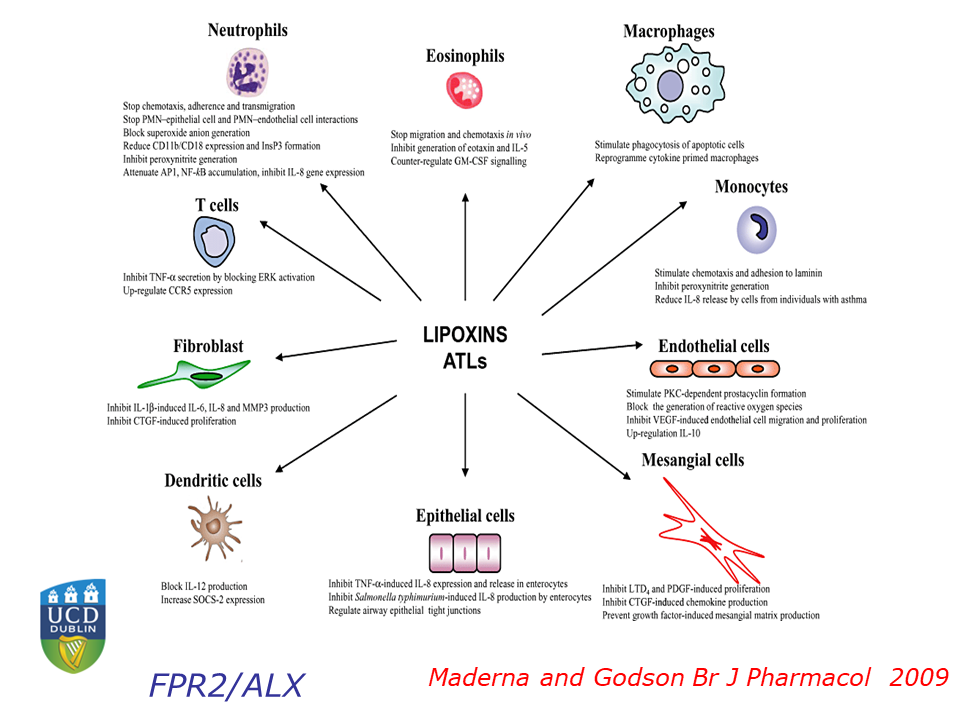
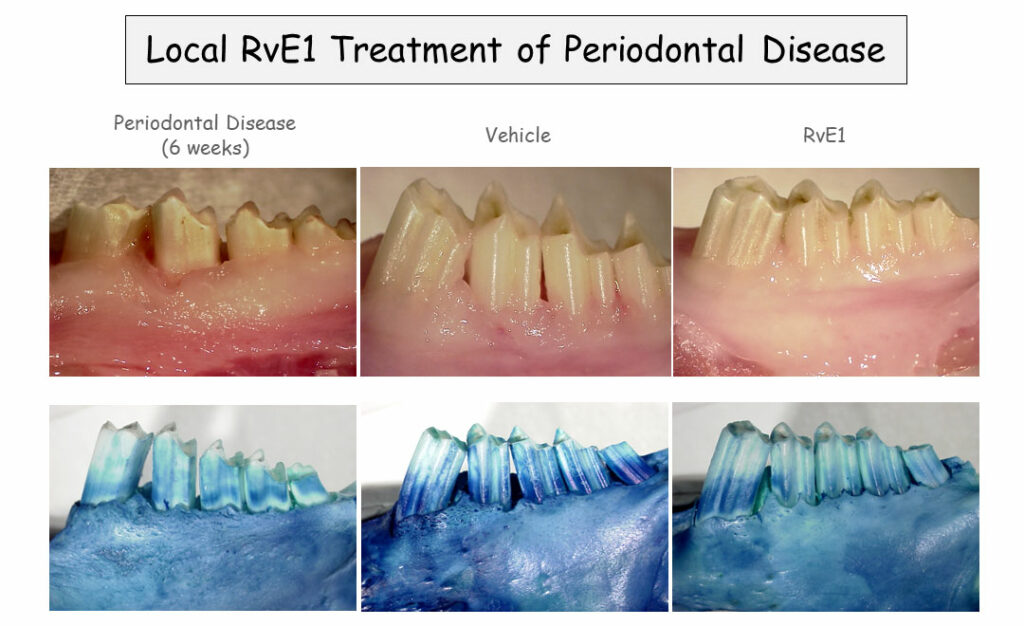

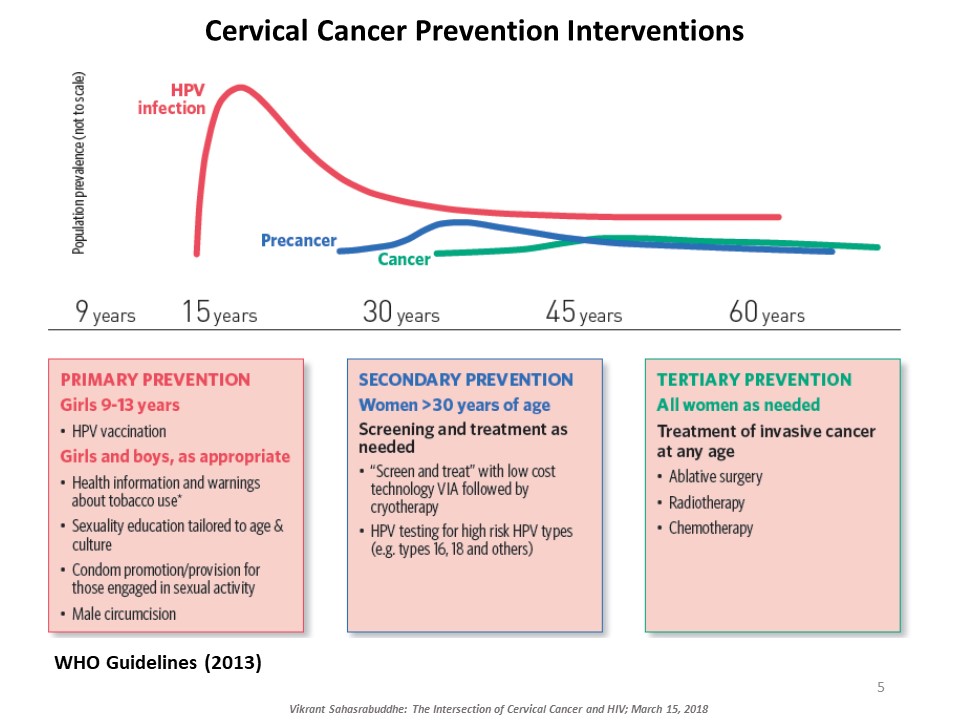
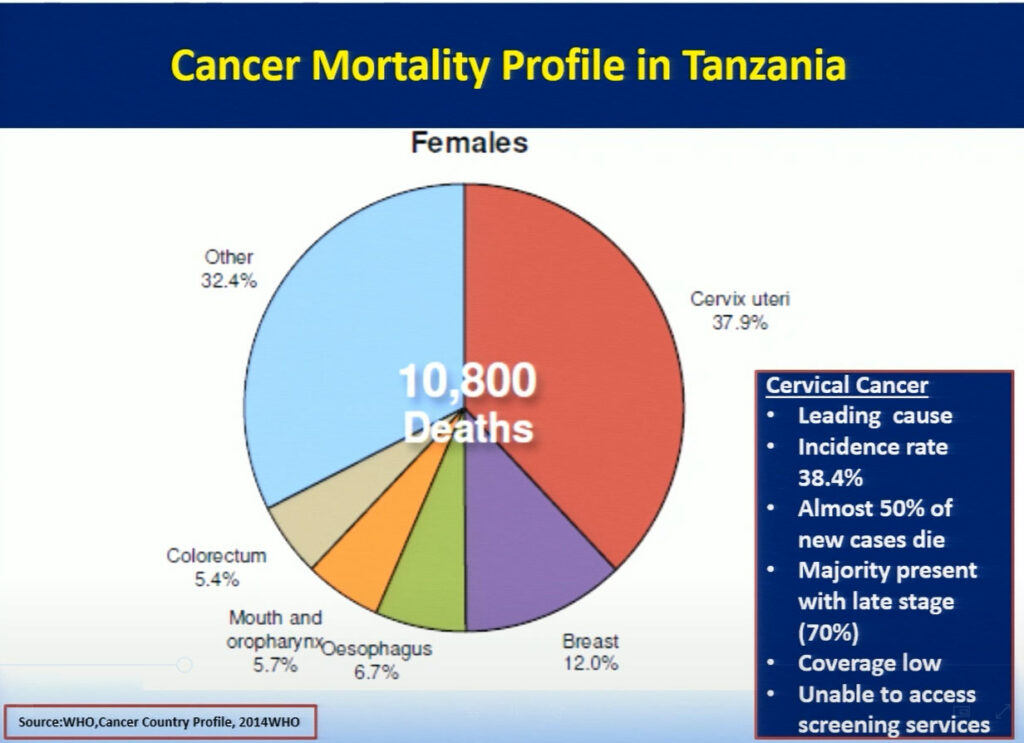
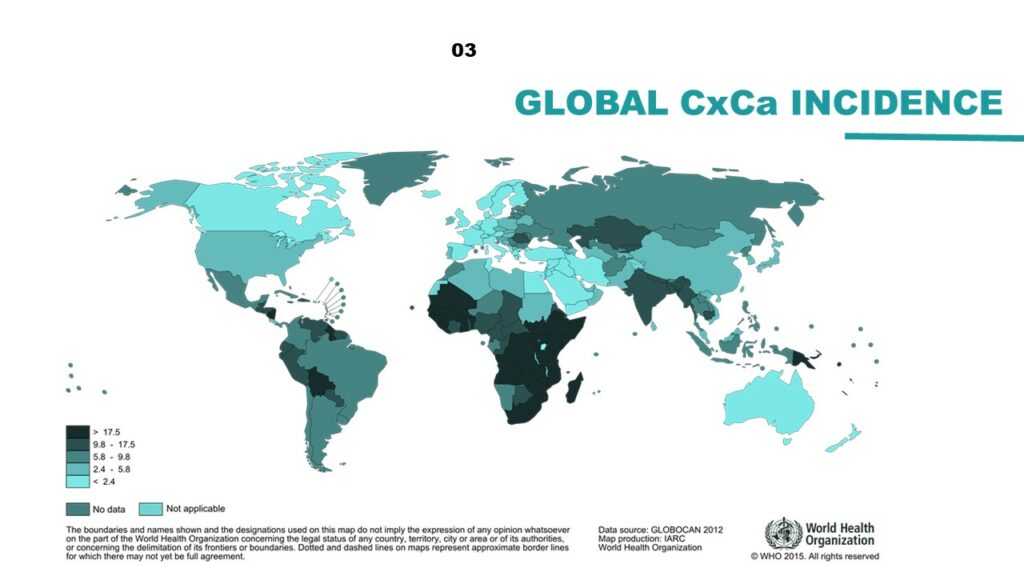
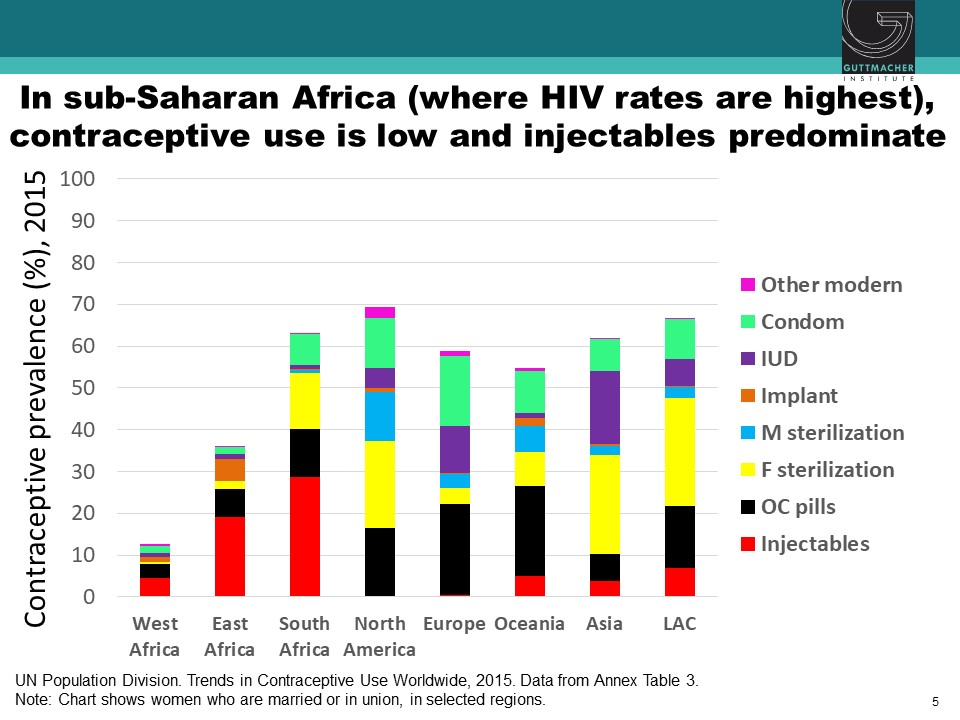














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}