The honorees, listed below, were each awarded US$100,000:
Physical Sciences & Engineering
Prof. Ido Kaminer, Technion – Israel Institute of Technology, 2021 Laureate
Prof. Guy Rothblum, Weizmann Institute of Science, 2020 Laureate (in absentia)
Chemistry
Prof. Rafal Klajn, Weizmann Institute of Science, 2021 Laureate
Prof. Emmanuel Levy, Weizmann Institute of Science, 2020 Laureate
Life Sciences
Prof. Yossi Yovel, Tel Aviv University, 2021 Laureate
Prof. Igor Ulitsky, Weizmann Institute of Science, 2020 Laureate (in absentia).
Israel’s newly-appointed President, Isaac Herzog, graced the ceremony with an appearance and a short speech. Herzog thanked Len Blavatnik for his philanthropy and support of scientific research, and praised scientists and their role in fighting COVID-19 in Israel, saying “Just as Pasteur’s experiments 150 years ago were the torch that illuminated the path to modern vaccines, the young scientists receiving the Blavatnik Awards tonight are illuminating the path to the future.”
Anchor of Israel TV’s Reshet 13, Dr. Hila Korach, served as Emcee. The President of the Israel Academy of Sciences and Humanities, Prof. Nili Cohen, gave opening remarks and introduced President Herzog. Afterward, The New York Academy of Sciences President and CEO, Nicholas B. Dirks, spoke about the importance of science to help humanity tackle the challenges ahead, and congratulated the Laureates.
Kfir Damari, Co-Founder of SpaceIL, was the keynote speaker and inspired the audience by sharing the story behind the inception of the Beresheet spacecraft and the creation of SpaceIL. Equally inspirational were Israeli Singer Marina Maximillian, youth performer Lia Schapira, and dancer Liron Ozery, who gave notable performances during the evening.
2020 and 2021 Laureates of the Blavatnik Awards in Israel. (L to R) Ido Kaminer, Rafal Klajn, Emmanuel Levy and Yossi Yovel.
VIP Guests at the event included:
Peter Thorén, Executive Vice President, Access Industries; Member of the Board of Governors, The New York Academy of Sciences
Avi Fischer, Chairman & CEO of Clal Industries
Uri Sivan, President of Technion – Israel Institute of Technology
Alon Chen, President of Weizmann Institute of Science
Ariel Porat, President of Tel Aviv University
Robert John Aumann, 2005 Nobel Laureate in Economics
Roger Kornberg, 2006 Nobel Laureate in Chemistry
Ambassador Neil Wigan, United Kingdom Ambassador to Israel
Ami Appelbaum, Chairman of Israel Innovation Authority;
Yulia Berkovich Shamalov, former Israeli politician
Ron Levkowitz, Chairman of First International Bank of Israel
To learn more about the Blavatnik Awards for Young Scientists, visit blavatnikawards.org.
Israel President Isaac Herzog.The New York Academy of Sciences President, Prof. Nicholas B. Dirks.Israel Academy of Sciences and Humanities President Prof. Nili Cohen.The 2021 Toast to Science. From left: Kfir Damari, Hila Korach, Nili Cohen, Peter Thorén, and Nick Dirks.“Science of Tomorrow” Panel Discussion. (L to R) Moti Segev, Oded Rechavi, Erez Berg, Tamar Ziegler.Israeli Singer and Songwriter, Marina Maximillian and youth performer Lia Schapira.Full-scale model of the Beresheet Spacecraft built by SpaceIL, on display at the Ceremony reception.
Growing up in Romania, Mircea Dincă’s was first exposed to science. Now he’s engineering an electric Lamborghini.
Published October 1, 2021
By Roger Torda
Mircea Dincă (left) poses with Nick Dirks, President and CEO of The New York Academy of Sciences.
Mircea Dincă creates materials in the lab with surface features that can’t be found in nature. He then makes variants with electrical properties that other scientists once thought impossible. This is groundbreaking basic research with many emerging applications. One is particularly exciting: a supercapacitor to power a Lamborghini supercar.
Dincă, a professor of chemistry at MIT, is this year’s Blavatnik National Awards for Young Scientists Laureate in Chemistry. He heads a lab that synthesizes novel organic-inorganic hybrid materials and manipulates their electrochemical and photophysical properties.
Dincă and his students work with metal-organic frameworks, or MOFs. “These are basically what I like to call sponges on steroids because they are enormously porous,” Dincă told the Academy in a recent interview. “They have fantastically high surface areas, higher than anything that humanity has ever known.”
Metal-Organic Frameworks (MOFs)
MOFs have a hollow, crystalline, cage-like structure, consisting of an array of metal ions surrounded by organic “linker” molecules. Scientists can “tune” their porosity, creating MOFs that can capture molecules of different properties and size.
To help conceptualize the large surface area of MOFs, Dincă says a gram of the material would, if flattened out, cover an entire football field. This means their pores can hold an almost unimaginably large number of molecules. One application capitalizing on this capacity is gas storage. For example, a canister filled with MOFs would hold nine times more CO2 than an empty canister. Other emerging uses have included devices to manage heat, antimicrobial products, gas separation, and devices for scrubbing emissions and carbon capture.
Dincă first encountered MOFs as a graduate student. Several years later, after considerable research on the electronic structure of materials, he started envisioning MOFs with properties that had not been widely considered before. “Previously, people thought that metal-organic frameworks are just ideal insulators,” Dincă said. “But we realized that there are certain types of building blocks that, when put together, would allow the free flow of electrical charges.” This was something of a paradigm shift in the field.
A Partnership with Lamborghini
Dincă and his students started synthesizing MOFs with a variety of organic ligands and metal combinations to create materials that are both porous and conducting. They also developed ways to grow MOF crystals so they can be more easily studied with imaging tools, permitting analysis of their structure, atom-by-atom. The new techniques and materials have led to MOFs that might prove valuable for batteries, fuel cells, and energy storage. Dincă’s lab and MIT have signed a partnership with Lamborghini to use MOF supercapcitors in the company’s planned Terzo Millennio sportscar.
Dincă and his students also study the use of MOFs as catalysts, and as chemical sensors. They explore how these materials interact with light, which could lead to smart windows that lighten or darken automatically. Better solar cells are yet another possible application.
More efficient air conditioning, with considerable environmental benefit, is another goal. Dincă has co-founded a start-up called Transaera to build MOF-based cooling equipment that pulls water molecules out of air so that the AC doesn’t work as hard. The key is tuning the pores of the MOFs to just the right size to capture water at just the right humidity.
Scaling up remains a challenge for many of these applications. “It’s one thing to make a few grams in a laboratory, it’s quite another to make hundreds of kilograms so you can take them out into the real world,” Dincă said.
“Thirsty for Knowledge”
Dincă grew up in Romania, and says he got his first taste of chemistry in 7th grade. An MIT departmental biography playfully suggests “that having a dedicated teacher that did spectacular demonstrations with relatively limited regard for safety” was the initial influence. One imagines awe-inspiring, semi-controlled explosions in the front of a classroom of 12 year olds. In the following years, Dincă started participating in the Chemistry Olympiads, and in 1998, when he was in high school, he won first place at an international competition in Russia.
At the time, Dincă found he was running up against limits to his education. “I think the biggest challenges to my becoming a scientist were, early on in Romania where I grew up, that we just didn’t have access to labs, to books,” Dincă said. “That made me thirsty for knowledge.” So Dincă was eager to travel to the U.S. when he was offered a scholarship for undergraduate studies at Princeton. He then earned a Ph.D. from UC Berkeley. He has been teaching and conducting research at MIT since 2008.
Dincă met his wife, who is also from Romania, while they were both students at Princeton. She is a lawyer, and the couple have two children, Amalia and Gruia. Dincă’s father is a retired Romanian Orthodox priest, and his mother, a retired kindergarten teacher.
When he is not with his family or at work, Dincă might be running, hiking, or taking photographs.
Constant Exposure to the Unknown
Dincă enjoys teaching, including freshmen chemistry. For his more advanced students and postdocs, Dincă says he fosters original thinking by giving them as much responsibility as possible. “As a Principal Investigator myself, I tend to be very hands-off,” Dincă explained. “And that’s good because it allows students to take ownership of their projects and become creative themselves. In fact, most of the best ideas in my lab come from the students, not myself.”
One of the best things about being a scientist, Dincă said, is constant exposure to the unknown, and he is pleased when his commitment to basic research is recognized. “Being a Blavatnik National Award Laureate is, of course, fantastic recognition of my research, of my group’s efforts,” Dincă said. “But also, most importantly for me, it is recognition of the fact that curiosity-driven research is still appreciated.”
While curiosity may drive Dincă’s scientific inquiries, he believes applied research with new classes of MOFs will help address important environmental challenges. At the same time, there can be no doubt that one application may prove especially thrilling. “Never in my wildest dreams did I believe that just thinking about electrical current in porous materials would take me on a path to helping make an electric Lamborghini,” Dincă said. “But that is where our research has led us.”
Andrea Alù is challenging the laws of physics to improve data transmission. Oh yeah, he’s working on an invisibility cloak, too!
Published October 1, 2021
By Roger Torda
Andrea Alù
Andrea Alù isn’t satisfied with how light waves and sound travel through objects and space. So he engineers new materials that appear to violate some well-established laws of physics. Enhanced wireless communication and computing technologies, improved bio-medical sensors, and invisibility cloaks are just some of the achievements of his lab.
“We create our own materials, engineered at the nanoscale,” explained Alù, who is Director of the Photonics Initiative at the Advanced Science Research Center at the City University of New York (CUNY). “We call them metamaterials, which push technologies forward, to realize optical properties, electromagnetic properties, or acoustic properties that go well beyond what nature and natural materials offer us.”
In a recent interview with The New York Academy of Sciences (the Academy), Alù explained a core behavior of light that is at the heart of his research:
One of the most basic phenomena in optics is light refraction, which describes the change in direction of propagation of an optical beam as it enters a material. We can understand this as the collective excitation of molecules and charges in the material, produced by light. In metamaterials, we make up our own molecules—we call them metamolecules.
Metamaterials feature many different geometries of at the nanoscale. Some can be engineered to interact with light in such a way that they may actually make objects disappear from sight. It is a phenomenon called “cloaking.” Alù continued:
Engineering at the Nanoscale
This engineering at the nanoscale allows us to change the ways in which light refracts as it enters a metamaterial. By bending light in unusual ways, we can actually realize highly unusual optical phenomena, like enhancing or suppressing the reflections and scattering of light from an interface, making a small object appear much larger, or conversely, even disappear altogether, by hiding it from the impinging electromagnetic waves.
“Invisibility” has long been part of our popular imagination and science fiction, from H.G. Wells’ novels to Star Trek and Harry Potter. A pioneering theoretical step dates back to 1968, when a Russian physicist wondered if a phenomenon called “negative refraction” might be possible. But no materials featuring this property were known, and some scientists believed none would be found because negative refraction might violate widely-used equations describing the propagation of light. Thirty years later, in 2000, a team of scientists was able to demonstrate negative refraction in a metamaterial for a certain frequency of electromagnetic radiation. A few years later, experiments demonstrated actual metamaterial cloaking, and Scientific American proclaimed: “Invisibility Cloak Sees Light of Day.”
Alù started working on metamaterials in 2002, when he spent a year at the University of Pennsylvania as a visiting student. He has conducted pioneering research in the field ever since. A major achievement came in 2013. Alù, then at the University of Texas at Austin, and his collaborators, demonstrated the cloaking of a three-dimensional object using radio waves. The work showed that antennas, like the ones in our cell phones, could be made transparent to radio-waves, a finding of potential commercial and military value, as it eliminates interference between closely-spaced transmitters.
A Childhood Fascination
Alù’s interest in light and other electromagnetic waves began as a child in Italy when he was fascinated by how our radios and television sets receive broadcast information without wiring. His interest intensified in high school when he realized a “beautiful common mathematical framework” describes the propagation of light, radio signals, and sound, and the fact that no information can be transmitted faster than the speed of light.
Alù went on to study at the University of Roma Tre, where he earned a Ph.D. in electronic engineering. After a postdoctoral fellowship at the University of Pennsylvania, he joined the faculty of UT Austin in 2009, and moved to CUNY in 2018.
Nanomaterials being developed in Alù’s lab may also improve near-field microscopy for better biomedical imaging, and lead to optical computers, enabling faster and more efficient PCs that use light instead of electric signals.
Yet another area of intense research for Alù and his research team has been “breaking reciprocity,” with implications for improved transmission of sound as well as radio waves and light. “Light, sound, and radio waves, typically travel with symmetry between two points in space,” Alù explained. “If you hear me, I can hear you back. If you can see me, typically you can see me back. This property is rooted into the time reversal symmetry of the wave equations.”
Connecting Basic and Applied Research
Alù said his lab’s work in breaking this symmetry with metamaterials is a good illustration of the connection between basic and applied research:
Interestingly, making materials that transmit waves one way and not the other started as a curiosity, but it has rapidly become extremely useful, from improving data rates with which our cell phones or WiFi technologies operate to protecting sensitive lasers from reflections. This has been a very exciting quest, from basic research to applications.
Alù began his research and teaching career in the U.S. only after he earned his Ph.D. in Italy and, as a result, he found he initially had a smaller professional network than many of his peers. But Alù says the U.S. was very welcoming, and he quickly caught up:
I come from Italy and I did all my undergraduate and graduate studies there. So, coming to the U.S. first as a postdoc, then as a faculty member, I didn’t have a large support network around me, I didn’t initially have a lot of connections…. But at the same time, I have to say, the United States offers tremendous opportunities, in particular to young scientists, to help build up their research groups, and to thrive.
Alù continued: “The U.S. is an amazing country in welcoming young people, new talent, and supporting them in the broadest possible terms… An excellent example of this is the Blavatnik National Awards program, and the broad range of scientists it recognizes.”
A married research duo are studying ways to better predict the feasibility and potential economic benefits of adopting battery technologies for renewable energy.
Published May 13, 2021
By Roger Torda
(Left to Right) Graham Elliott and Shirley Meng at the 2019 Blavatnik National Awards Ceremony at the American Museum of Natural History
What can we learn from a marriage of physical and social sciences?
In an intriguing collaboration, they developed ways to better predict the feasibility and potential economic benefits of adopting battery technologies to integrate renewable energy, such as solar and wind energy, into energy grids. Together with their research team members, they published “Combined Economic and Technological Evaluation of Battery Energy Storage for Grid Applications” in the journal Nature Energy.
Meng is the Zable Chair Professor in Energy Technologies and Director of the Institute for Materials Design and Discovery at the University of California San Diego (UCSD). Elliott is also at UCSD, where he is Professor and Chair of the Department of Economics. We recently interviewed both to discuss this collaboration and what they learned through the process.
Can you tell us how this collaboration was initiated?
Meng: UCSD is a place where interdisciplinary and convergent research is not only highly valued but practiced. I founded the Sustainable Power and Energy Center (SPEC) at UCSD in 2015. SPEC reaches out beyond engineering and physical sciences to study economic and sociological issues that need to be addressed to create truly robust ecosystems for low-carbon electric vehicles and carbon-neutral microgrids. We won a competitive grant from the US Department of Energy, which provided the resources for this work.
Why did you choose to study batteries for energy grid applications? What question about batteries did you study?
Meng: With energy grids showing their age and continuing to distribute energy generated with high environmental costs, efforts that enable grids to distribute cleaner, renewable energy more efficiently would be a technological advance with a positive societal impact. While there have been exciting moves toward renewables, many problems lie ahead if we are to move from renewables being important to renewables being dominant.
Elliott: Grid energy storage remains a major challenge both scientifically and economically. Batteries, or energy storage systems, play critical roles in the successful operation of energy grids by better matching the energy supply with demand and by providing services that help grids function. They will not just transform the market for supplying energy but also transform consumer demand by lowering the prices of energy for households and businesses.
In this work, we studied the potential revenues that different battery technologies deployed in the grid will generate through models that consider market rules, realistic market prices for services, and the energy and power constraints of the batteries under real-world applications.
Bringing these together in an interactive way—examining the engineering and economic aspects as two parts of the problem together—allows for a complete look at the problem, and ultimately a better outcome for the economy.
Graham Elliott
What was the biggest finding of this collaboration? Were you surprised by your findings?
Meng: We found that while some battery technologies hold the greatest potential from an engineering perspective, the choice based on economics is less clear. The current rules of grid operations dictate which battery technologies are used for those particular grids—some of these rules may be out-of-date, and will be updated as the grids modernize. So even though we continue to see improvement in the energy/power performance of battery technologies and reduction in cost, policymakers are the ultimate decision-makers. Policymakers setting those rules have considerable influence on how fast and how successfully those battery technologies can be deployed, and therefore industry needs to work closely with policymakers to define the best practices for faster deployment of battery technologies.
We also found that there are a wide variety of factors that should be considered in choosing a battery technology. For instance, the battery recycling method is an important technical variable that determines the sustainability of a particular battery technology.
How could your findings eventually affect individual people and society? How can it help our economy?
Elliott: All gains in human welfare arise from what economists call productivity gains—people creating more with less effort, so there is more to go around. Technological advances in energy storage enable productivity gains. But for it to work, we need not only to be able to provide effective energy storage from an engineering perspective, but also it needs to be economically feasible. Different choices at the engineering stage mean differences in the economic feasibility, and how markets are arranged impacts engineering choices. Bringing these together in an interactive way—examining the engineering and economic aspects as two parts of the problem together—allows for a complete look at the problem, and ultimately a better outcome for the economy.
Meng: We are delighted to see to see that battery grid storage is starting to gain more momentum—policymakers are becoming informed about both economic and scientific, and engineering aspects of battery technologies.
A small-scale energy grid at the University of California San Diego, consisting of a network of solar cells with battery storage (Credit: University of California San Diego)
What did you learn from this collaboration? Are there any tips you would like to share with other researchers who would like to pursue similar collaborations between physical and social sciences?
Meng: Perhaps the most important thing for the collaborative team to do is to build a common vocabulary so we can truly understand each other. In our case, we started by explaining the most basic symbols and units in engineering, like the energy unit Wh (Watt-hour) and the power unit W (Watt). Without understanding the differences between these symbols, we will make mistakes in constructing important parameters in our economic modeling.
Elliott: Another thing we learned is that different fields have very different understandings of the big picture. Collaboration across fields helps focus everyone’s efforts. For example, engineers typically view markets as fixed, and the engineering problem is to find something that works for the market. Economists tend to think of products (such as batteries) as fixed and design markets that work for the available products.
There is a whole research area waiting patiently for economists to understand which parts of the engineering problem are important and for scientists and engineers to understand from their perspective which parts of the market design are important.
Early-career scientist, outstanding senior scientist each to receive US$200,000 in program sponsored by Takeda Pharmaceuticals
New York, NY | April 14, 2021 – The New York Academy of Sciences (NYAS) has opened nominations for the 2022 Innovators in Science Award, which will recognize significant achievement among early-career and senior scientists in the field of gastroenterology. This marks the first time scientists engaged in transformative research in gastroenterology will be eligible for the award, administered by the Academy and sponsored by Takeda Pharmaceuticals.
The program accepts nominations from eligible research institutions around the world to recognize the work of a promising early-career scientist and an outstanding senior scientist. Winners in each category will receive an unrestricted award of US$200,000 for having distinguished themselves for the creativity and impact of their research.
The Academy is accepting nominations through May 27, 2021, from more than 400 international universities and academic institutions, select government-affiliated and non-profit research institutions and the program’s Scientific Advisory Council, composed of renowned science and technology leaders. Candidates must be nominated by their institution and may not be self-nominated.
A judging panel composed of scientists, clinicians and international experts in gastroenterology will determine the two winners based on the quality, impact, novelty and promise of their research. They will be announced in January and honored at the 2022 Innovators in Science Award ceremony and symposium, scheduled for March 28-29, 2022, in Tokyo, Japan, as health and travel conditions allow.
“After one of the most challenging years of our time, recognizing and celebrating advancements in science is more important than ever,” said Nicholas B. Dirks, President and CEO of The New York Academy of Sciences. “The world is seeing firsthand how innovative science and thinking can improve human health, and we are committed to honoring those who are leading the way. The Innovators in Science Award salutes ground-breaking researchers who have developed science-based solutions to debilitating diseases, improving quality of life for people all over the world.”
Since its inception, the Innovators in Science Award has focused on acknowledging outstanding research and contributions in fields of medicine aligned with Takeda’s core therapeutic areas. The inaugural award recognized neuroscience discovery, followed the next year by regenerative medicine, rare disease research in 2020 and the latest on research in gastrointestinal and liver diseases. Recent research shows that 20-40% of adults worldwide are affected by at least one functional gastrointestinal disorder, which can dramatically impact quality of life.
Nominations may be submitted by representatives from the nominating institution through the Innovators in Science Award website via its online submission platform: https://innovatorsinscienceaward.smapply.io. Please refer to the guidelines and FAQ sections for other details on eligibility, nomination materials and the selection process.
Shruti Puri, PhD, helps explain the challenges and the potential computational power this exciting new technology may bring about.
Published March 22, 2021
By Liang Dong, PhD
Shruti Puri, PhD, Yale University
Quantum computing is a radically new way to store and process information based on the principles of quantum mechanics. While conventional computers store information in binary “bits” that are either 0s or 1s, quantum computers store information in quantum bits, or qubits. A qubit can be both 0 and 1 at the same time, and a series of qubits together remember many different things simultaneously.
Everyone agrees on the huge computational power this technology may bring about, but why are we still not there yet? To understand the challenges in this field and its potential solutions, we recently interviewed Shruti Puri, PhD, who works at the frontier of this exciting field. Puri is an Assistant Professor in the Department of Applied Physics at Yale University, and a Physical Sciences & Engineering Finalist of the 2020 Blavatnik Regional Awards for Young Scientists, recognized for her remarkable theoretical discoveries in quantum error correction that may pave the way for robust quantum computing technologies.
What is the main challenge you are addressing in quantum computing?
Thanks to recent advances in research and development, there are already small to mid-sized quantum computers made available by big companies. But these quantum computers have not been able to implement any practical applications such as drug and materials discovery. The reason is that quantum computers at this moment are extremely fragile, and even very small noise from their working environment can very quickly destroy the delicate quantum states. As it is almost impossible to completely isolate the quantum states from the environment, we need a way to correct quantum states before they are destroyed.
At a first glance, quantum error correction seems impossible. Due to the measurement principle of quantum mechanics, we cannot directly probe a quantum state to check if there was an error in it or not, because such operations will destroy the quantum state itself.
Fortunately, in the 1990s, people found indirect ways to faithfully detect and correct errors in quantum states. They are, however, at a cost of large resource overheads. If one qubit is affected by noise, we have to use at least five additional qubits to correct this error. The more errors we want to correct, the larger number of additional qubits it will consume. A lot of research efforts, including my own, are devoted to improving quantum error correction techniques.
What is your discovery? How will this discovery help solve the challenge you mention above?
In recent years, I have been interested in new qubit designs that have some in-built protection against noise. In particular, I developed the “Kerr-cat” qubit, in which one type of quantum error is automatically suppressed by design. This reduces the total number of quantum errors by half! So, quantum computers that adopt Kerr-cat require far fewer physical qubits for error correction than the other quantum computers.
Kerr-cat is not the only qubit with this property, but what makes the Kerr-cat special is that it is possible to maintain this protection while a user tries to modify the quantum state in a certain non-trivial way. As a comparison, for ordinary qubits, the act of the user modifying the state automatically destroys the protection. Since its discovery, the Kerr-cat has generated a lot of interest in the community and opened up a new direction for quantum error correction.
As a theoretician, do you collaborate with experimentalists? How are these synergized efforts helping you?
Yes, I do collaborate quite closely with experimentalists. The synergy between experiments and theory is crucial for solving the practical challenges facing quantum information science. Sometimes an experimental observation or breakthrough will provide a new tool for a theorist with which they can explore or model new quantum effects. Other times, a new theoretical prediction will drive experimental progress.
At Yale, I have the privilege to work next to the theoretical group of Steve Girvin and the experimental groups of Michel Devoret and Rob Schoelkopf, who are world leaders in superconducting quantum information processing. The theoretical development of the Kerr-cat qubit was actually a result of trying to undo a bug in the experiment. Members of Michel’s group also contributed to the development of this theory. What is more, Michel’s group first experimentally demonstrated the Kerr-cat qubit. It was just an amazing feeling to see this theory come to life in the lab!
Are there any other experimental developments that you are excited about?
I am very excited about a new generation of qubits that are being developed in several other academic groups, which have some inherent protection against noise. Kerr-cat is one of them, along with Gottesman-Kitaev-Preskill qubit, cat-codes, binomial codes, 0−π qubit, etc. Several of these designs were developed by theorists in the early 2000s, and were not considered to be practical. But with experimental progress, these have now been demonstrated and are serious contenders for practical quantum information processing. In the coming years, the field of quantum error correction is going to be strongly influenced by the capabilities that will be enabled by these new qubit designs. So, I really look forward to learning how the experiments progress.
Professor Pardis Sabeti was able to apply findings from her research on Ebola to now develop a test for detecting COVID-19.
Published March 9, 2021
By Brittany Aguilar, PhD
Pardis Sabeti, MD, DPhil, MSc
This isn’t the first time that Pardis Sabeti, MD, DPhil, MSc, a professor of organismic and evolutionary biology at Harvard University, and newly elected member of the National Academy of Medicine, has worn the hat of viral genome detective in the earliest days of a deadly outbreak or viral disease. Sabeti and her team began sequencing Ebola samples just days after the virus was first detected in Sierra Leone during the 2013-2016 West African outbreak. Since January 2020, she has been working on diagnostics for COVID-19, developing models to predict the most sensitive and accurate assay design candidates for the rapid detection of SARS-CoV-2, including an assay that harnesses the powerful accuracy of CRISPR technology.
Describe the innovative, rapid COVID-19 test that you helped create—how does it work, and why is it an improvement on current testing methods?
Over the last several years, my lab, colleagues, and I have been developing an assortment of technologies for genomic surveillance of pathogens. In particular, we have been deeply invested in CRISPR technologies. CRISPR was first discovered within bacterial immune systems, where it is used to protect the bacteria from invading pathogens by rapidly identifying and targeting a genomic sequence with very high fidelity. Thus, it is immensely powerful as a diagnostic tool, since it can be designed to detect any sequence of genetic material with impressive accuracy.
It is an incredibly exciting technology: it is highly accurate, it would be able to rapidly detect pathogens using little equipment and a simple, paper-strip read-out, and it could be developed in a matter of days to detect newly discovered pathogens or new variants of known pathogens. Crucially, the test is also inexpensive to manufacture, which means it could be easily scaled and distributed as pathogens—or novel variants of pathogens—emerge.
Throughout the duration of the COVID-19 pandemic, some have suggested that testing is optional, unnecessary or unreliable—can you describe why the creation of rapid, reliable tests is so important? Does that change depending on where we are in the infection curve?
Testing is extremely critical to fighting the spread of any infectious disease, and this has been demonstrated through history. However, testing technology has been achievable but not prioritized—if we had invested in this space after the SARS-CoV epidemic [the SARS outbreak in 2003], I believe we could have been poised to respond to SARS-CoV-2 before it spread throughout the world.
The need for diagnostics is critical everywhere, from pre-empting a pandemic, to response and recovery. To be as useful as possible, diagnostics must also be affordable and accessible to all—this is not just in infectious disease but throughout all medicine. The sooner individuals and communities have information, the better they can respond, enabling better outcomes.
You wrote a book last year entitled “Outbreak Culture.” Are there any key learnings from that book that can be applied to COVID or future pandemics?
In this book we argue that a dysfunctional “outbreak culture”—the collective mindset that develops among responders and communities that emerges in the chaos and crucible that is disease outbreaks—poses a great threat to our ability to curb outbreaks and save lives, and that we must continually watch for and dismantle toxic response systems where possible. This includes the data and resource hoarding, perverse capitalistic incentives, the spread of misinformation, and the loss of empathy and good citizenship.
I think people are still just beginning to understand the gravity of outbreak culture and how it is operating amidst COVID. For example, we all now know the importance of detecting outbreaks, through track-and-trace methods, before they have the chance to spread widely. But what is given less attention is how those efforts can be sidelined or undermined by many surrounding societal and political forces.
I always advocate for a massively increased effort for empathy during outbreaks. We need resilient communities to be able to do the best work against infectious disease. With our trust in our fellow citizens, our leaders, and our scientists undermined during this time, it is crucial to work within the community and low to the ground. We must listen to others, respect their opinions, and understand their fears. For that reason, I believe we must double down on empathy when it comes to community participation. If we do not work with communities and support them in the right ways, we end up causing more harm than good.
About Prof. Sabeti
Pardis Sabeti, MD, DPhil, MSc is a Professor at the Center for Systems Biology and Department of Organismic and Evolutionary Biology at Harvard University and the Department of Immunology and Infectious Disease at the Harvard School of Public Health. She was a 2016 and 2017 Finalist for the Academy’s Blavatnik National Award for Young Scientists. To learn more about Dr. Sabeti and her work, click here to listen to the “Deciphering Zika” podcast.
By definition, a rare disease is one that afflicts relatively few people compared to the general population. Collectively, though, there are over 7,000 of these conditions known, causing immense suffering for an estimated 300 million patients. Because most rare diseases stem from specific genetic mutations, they’ve proven difficult to treat.
Genome sequencing and molecular medicine might soon change those grim statistics, though. For example, using short DNA or RNA sequences complementary to the messenger RNA for a gene, researchers can inhibit the expression of the associated protein. These complementary sequences—called antisense oligonucleotides—could soon be delivered as drugs to treat many rare diseases.
On October 2, 2020, The New York Academy of Sciences and Takeda Pharmaceuticals hosted the Frontiers in Rare Diseases: 2020 Innovators in Science Award Symposium, an event highlighting breakthroughs in rare diseases research and honoring 2020 Innovators in Science Award Winners Adrian Krainer, PhD and Jeong Ho Lee, MD, PhD. Presentations, a panel discussion, and a virtual poster session covered the basic science, recent clinical breakthroughs, and remaining challenges in this rapidly evolving field.
Symposium Highlights
While many rare diseases are inherited, others arise through mutations in somatic cells during life.
Antisense oligonucleotides can alter the expression of specific genes, potentially mitigating or reversing many genetic diseases.
Clinical trials for rare disease therapies must be tailored to the pathogenesis of each disease.
Redirecting neural stem cells to become neurons could treat many neurodegenerative diseases.
The COVID-19 pandemic is inspiring new collaborations that could be adapted to rare disease research.
Speakers
Jeong Ho Lee, MD, PhD Korea Advanced Institute of Science and Technology
Adrian Krainer, PhD Cold Spring Harbor Laboratory
Annemieke Aartsma-Rus, PhD Leiden University Medical Center
Don Cleveland, MD, PhD University of California, San Diego
Huda Zoghbi, MD Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital
Brad Margus Cerevance
Graciana Diez-Roux, PhD Telethon Institute of Genetics and Medicine
David Fajgenbaum, MD University of Pennsylvania
Anne Heatherington, PhD Takeda Pharmaceuticals
Sponsors
The Winner’s Circle
Speakers
Jeong Ho Lee, MD, PhD Korea Advanced Institute of Science and Technology
Adrian Krainer, PhD Cold Spring Harbor Laboratory
Not Born This Way
Jeong Ho Lee, Early-Career Scientist winner of the 2020 Innovators in Science Award, discussed his work studying how somatic cell mutations—mutations that occur after development, during the normal process of cell division—result in rare neurological diseases caused by somatic cell mutations in the brain. Much of the recent boom in work on genetic diseases has focused on germline mutations. Because these mutations occur early in embryonic development, they show up in many types of cells throughout the body and are passed on to future offspring. These rare germline mutations can often be identified by sequencing the genomes of cells in easily accessible tissues, such as blood or skin. With advances in next-generation sequencing, “it’s become much easier to identify the germline mutations coding for many rare neurological disorders,” said Lee.
Somatic cell mutations occur throughout life, in every part of the body.
Nonetheless, germline mutations account for only a minority of rare neurological disorders. For example, 98% of epilepsy cases cannot be explained by germline mutations.
“We hypothesized that somatic cell mutations may be responsible for these unexplained neurological [diseases],” said Lee.
Somatic cell mutations occur during the ordinary cell division process that takes place billions of times in developing embryos, and continues to occur throughout life as somatic, or non-gamete, cells turn over.
DNA replication isn’t perfect, and human cells average 0.1 to 3 mutations per genome every time they divide. Lee theorized that a patient who hadn’t inherited an epilepsy-causing germline mutation might instead acquire somatic cell mutations in a subset of brain cells during development or later in life. If that happened, the mutation would only show up in the affected area of the brain, not in any other cells of the body.
One treatment for certain types of epilepsy is to resect the portion of the brain causing the seizures. Lee and his colleagues took samples of the brain tissue resected in these operations, along with blood samples from the same patients, and performed deep DNA sequencing to identify somatic mutations that occurred only in the affected brain tissue, not in the blood. They identified such mutations, including ones unique to genes involved in motor nerve activity, in 30% of the patients. When the scientists introduced the same mutations into a small percentage of neurons in developing mice, the animals developed epilepsy.
Next, the investigators looked at brain tumors, which can also cause epilepsy. One rare brain tumor type involves both glial cells and neurons, triggering epilepsy. Sequencing genetic information from cells in the tumors revealed that in 46% of affected patients, the glial cells and the neurons in the tumor shared an identical mutation.
“It means that the…neural stem cell already contained this… mutation,” said Lee, “and differentiated into the neuron and the glial cell.”
That could explain the high rate of disease recurrence in patients with these tumors; even if surgeons remove the entire tumor, the mutant stem cells might continue to produce more defective neurons and glial cells, which could then seed the growth of a new tumor.
To confirm that, Lee’s team collected an additional round of samples, this time sequencing cells not only from resected brain tumors and blood, but also from the subventricular zone in each patient’s brain, an area rich in undifferentiated neural stem cells. They found the tumor-associated mutation in the subventricular zone samples as well as the tumors, indicating that the error occurred in the neural stem cells, whose neuronal and glial descendants then migrated to where the tumor grew.
The researchers are also looking at neurodegenerative disorders such as Alzheimer’s disease.
“We hypothesized that somehow brain somatic mutation maybe accumulates over aging, and maybe associates with [Alzheimer’s disease development],” said Lee.
By performing deep sequencing on brain tissues from patients with and without Alzheimer’s disease, he and his colleagues identified somatic mutations unique to the diseased brains, supporting their theory.
In addition to identifying the underlying mechanisms behind neurological diseases, Lee is trying to help patients in other ways. In one effort, he has begun providing his results to clinicians to use in genetic counseling. Because conditions caused by somatic mutations aren’t heritable, while those caused by germline mutations are, patients who might be considering having children need to know which category they’re in.
The investigators are also trying to find ways to repair or mitigate the effects of somatic mutations in the brain, but it’s a tall order.
“Even if we found a molecular genetically validated target in the patient’s brain, it would be very difficult to develop a traditional drug to penetrate the blood-brain barrier and regulate the target,” Lee explained.
Instead, he’s hopeful that chemically modified strings of nucleic acids, called antisense oligonucleotides, will be able to target the somatic mutations he’s identified.
“I believe in the next five, ten, or twenty years, we probably can solve a lot of the rare neurological disorders,” he said.
Different Diseases, Different RNA Splices
Adrian Krainer won the 2020 Innovators in Science Senior Scientist Award, recognizing years of work spent developing treatments for rare diseases. Krainer and his colleagues were the first to develop an effective drug to treat spinal muscular atrophy. Affecting about 1 in 10,000 people worldwide, spinal muscular atrophy is an inherited genetic disease caused by a defect in the SMN1 gene. SMN1 encodes the SMN protein, which is essential for motor neuron survival. Patients with the mutation experience progressive loss of motor neurons, leading to loss of muscle control and, in most forms of the disease, early death.
Another gene, SMN2, also encodes the SMN protein, but cells usually splice out one of the protein coding sequences, or exons, from the SMN2 messenger RNA, preventing it from making the full-length protein. As a result, 80-90% of the protein translated from SMN2 RNA is truncated, nonfunctional, and rapidly degraded by the cell. Krainer reasoned that preventing the exon-skipping event might allow patients’ unmutated SMN2 genes to produce more functional SMN protein, overcoming the deficit caused by their mutated SMN1 genes. To do that, his team turned to antisense oligonucleotides, which encode the complementary, or antisense, sequence of a specific RNA target. When introduced into a cell, the antisense oligonucleotide binds specifically to its target sequence, triggering various cellular responses.
By designing an antisense oligonucleotide that altered the splicing of SMN2 messenger RNA, the researchers were able to get SMN1-mutant cells to produce more functional SMN protein. Subsequent preclinical and clinical trials proved that their antisense oligonucleotide also works in spinal muscular atrophy mouse models and in patients, respectively, significantly mitigating their motor neuron losses.
“Therefore this is a way that allows them to make closer to normal levels of a functional SMN protein in the presence of this drug,” said Krainer.
The oligonucleotide, now sold as nusinersen (Spinraza), was approved in the US in 2016 and the EU in 2017. Over 11,000 patients now receive it worldwide.
Nusinersen (Spinraza) pioneered many aspects of molecular medicine.
Based on the success of nusinersen, Krainer and his colleagues have begun looking at other RNA processing events to target with antisense oligonucleotides. One project focuses on familial dysautonomia, an inherited genetic disorder that affects only 310 known patients worldwide. These individuals have profound defects in their sensory neurons and autonomic nervous system, leading to symptoms that range from insensitivity to pain to difficulty swallowing.
“It is a very severe disease, a rare disease with a complex set of symptoms,” said Krainer, adding that “median survival is about 40 years of age.”
The condition is caused by a mutation in the gene for a protein called ELP1. As in SMN2, the mutation causes one exon of the gene’s messenger RNA to be spliced out, leading to a loss of functional ELP1 protein.
“So, we started targeting this aberrant splicing event using the same screening strategy and the same chemistry that we used…for spinal muscular atrophy,” said Krainer.
That effort identified an antisense oligonucleotide that can reverse the ELP1 RNA splicing defect in cultured cells from patients, as well as a transgenic mouse model.
“We feel that this is ready for clinical development; it is a challenge, though, because of the rarity of this disease,” said Krainer.
With only a few hundred patients in the US and Israel, the market for familial dysautonomia therapies is minuscule, and effective screening of potential carriers of the affected gene has led to very few new patients being born.
Not all RNA splicing-related diseases are rare, though. Work by several researchers has shown that in at least some cases, a change in the splicing of messenger RNA can help cancer cells grow. Alternatively, spliced forms of the messenger RNA for the PKM gene can produce two different isoforms of the metabolic enzyme pyruvate kinase. PKM1 predominates in normal adult tissues, while tumors and some developing tissues favor PKM2 production.
Using the same approach that worked in their rare disease work, Krainer’s team screened antisense oligonucleotides and identified candidates that bound the PKM messenger RNA and directed its splicing to favor PKM1 protein production. Putting these oligonucleotides into hepatocellular carcinoma cells causes the cells to shift their metabolism and slow their growth. In a mouse model of hepatocellular carcinoma, injecting the antisense oligonucleotides led to a significant reduction in tumor growth compared to control animals treated with saline solution.
Annemieke Aartsma-Rus, PhD Leiden University Medical Center
Don Cleveland, MD, PhD University of California, San Diego
The Kindest Cut
Annemieke Aartsma-Rus began the meeting’s third session with a presentation about her group’s efforts to address Duchenne muscular dystrophy with antisense oligonucleotides. While Krainer’s approach to rare diseases focuses on conditions where an exon needs to be added back into a messenger RNA, Aartsma-Rus described a case where it’s better to remove one.
Duchenne muscular dystrophy is an X-linked genetic disorder. In most cases, a mutation in the dystrophin gene shifts the messenger RNA’s reading frame, causing translation of the dystrophin protein to fail.
“Patients become wheelchair dependent around the age of 12, need assisted ventilation around the age of 20, and generally die in the second to fourth decade of life,” said Aartsma-Rus.
A related but milder disorder, Becker muscular dystrophy, also involves a deletion in the dystrophin gene but doesn’t shift the messenger RNA’s reading frame. As a result, patients with Becker muscular dystrophy produce partially functional dystrophin and exhibit a slower disease progression.
Skipping an exon in the RNA can fix a frame-shift mutation.
Looking at the affected DNA and RNA sequences, Aartsma-Rus reasoned that most Duchenne muscular dystrophy patients could make Becker-like dystrophin, if their cells could simply skip the affected exon in their dystrophin messenger RNA. To test that, she and her colleagues developed chemically modified antisense oligonucleotides that would remain stable in blood and tissues, and began testing them as potential drugs. By designing an oligonucleotide that targeted RNA splicing, the team restored dystrophin expression in cultured cells carrying a Duchenne muscular dystrophy mutation.
The researchers discovered a potential roadblock in a mouse model: antisense oligonucleotides injected into the animals’ tail veins were absorbed almost entirely by the liver and kidneys. The investigators could inject the molecules directly into muscles instead, but that clearly wouldn’t be a practical way to deliver treatment to patients.
“We have over 700 different muscles, and you’ll have to treat patients repeatedly,” said Aartsma-Rus, “so local injection of each and every muscle weekly or even monthly is likely not realistic.”
However, in a mouse model of Duchenne muscular dystrophy, the team discovered that oligonucleotides injected into the animals’ tail veins were absorbed into muscles ten times better than they had been in wild-type mice.
“The first time we thought we’d made a mistake, so we repeated it a couple of times,” said Aartsma-Rus, “but every time we saw that there was higher uptake by the dystrophic muscle than the healthy muscle.”
Dystrophin deficiency causes muscle cells to become more permeable, leading to leakage of cellular components, but this leakage works in both directions; the dystrophic cells readily absorbed oligonucleotides that healthy cells excluded.
Flush with this preclinical victory, the team began setting up clinical trials in 2007. The initial multi-center, open-label trial found that the antisense oligonucleotides caused no serious side effects, and eight of the twelve patients tested saw their conditions remain stable throughout the trial. To evaluate efficacy, the investigators moved into a phase 2b trial, which continued to show dose-dependent effectiveness in treated patients. However, a larger phase 3 trial yielded disappointment, with no significant difference in outcomes between treated and control patients.
“So, what happened [to explain why] we see these beneficial effects in the phase 2 trial, but in the phase 3 trial we see no effect?” Aartsma-Rus asked.
Analyzing the results and the disease further, she realized that the trials were built on the flawed assumption that the patients’ progression would be linear. Instead, they realized that younger patients tend to remain stable for an extended period, followed by a rapid decline later in life. By mixing different ages in the phase 3 trial, patients with worse disease symptoms likely masked any treatment benefits in those with milder symptoms.
Looking at the trial’s failure, Aartsma-Rus concluded that she and her colleagues should have opened discussions with regulatory agencies sooner, and studied the natural history of the disease more thoroughly, before initiating the phase 3 study. Unfortunately, the expensive late-stage failure has soured companies on further clinical development of exon-skipping antisense oligonucleotides for Duchenne muscular dystrophy. Aartsma-Rus has since focused on preventing such an outcome in the future.
“Now we have an open dialogue with academics, with patients, with regulators in the EU, and it is also starting in the US, developing new outcome measures,” she said, adding that “future trials will be better.”
Batting for Lou Gehrig
Don Cleveland discussed his group’s efforts to treat neurodegenerative diseases in the brain, especially those that develop gradually with age. In many of these conditions, such as Alzheimer’s and Parkinson’s disease and amyotrophic lateral sclerosis, “the genes that contribute to disease are all widely expressed…throughout the nervous system, not within individual classes of neurons,” said Cleveland. Mechanistic studies have suggested that decreasing the expression of the defective gene products in some of these cells could moderate the course of disease, so Cleveland and his colleagues set out to do just that.
The researchers first focused on amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease. About four to five million people alive today will die of ALS, a progressive neurodegenerative condition that can be inherited or occur spontaneously in adults. One inherited form of the disease stems from a mutation in the gene for superoxide dismutase, which causes neurons to die through mechanisms that aren’t entirely clear yet.
Using the same strategy as his co-speakers, Cleveland’s lab designed antisense oligonucleotides that bind specifically to the superoxide dismutase messenger RNA and target it for degradation in the cell. That decreases the level of the enzyme, an intervention that had previously been shown to ameliorate ALS progression in a mouse model of the disease. The next challenge was delivering the oligonucleotides to affected neurons in the brain.
Antisense oligonucleotides have immense potential to be used as drugs against a wide range of diseases.
“These DNA drugs were ten to fifteen times the size of a typical drug, and they’re heavily charged,” said Cleveland, “so the pharmacology textbooks all said that there was no uptake mechanism that would permit them to be efficiently taken up [by neurons].” Nonetheless, he continued, “we tried it anyway, and it turns out that the cells of the nervous system hadn’t read the textbooks.”
Injecting the antisense oligonucleotides into the cerebrospinal fluid of mice genetically modified to develop severe ALS doubled the animals’ survival times.
While the superoxide dismutase defect was the first ALS mutation discovered, the most common cause of the inherited form of the disease is a mutation in a gene called ORF72, which inserts extra nucleotides into a non-coding region of the gene. This causes defective messenger RNA to accumulate, killing neurons because of a lack of functional ORF72 gene products and the accumulation of toxic byproducts of the altered gene. Antisense oligonucleotides targeting the defective RNA, however, inhibit its accumulation without reducing the production of working ORF72 gene products in cultured cells.
In an animal model of the ORF72 defect, the results were even more impressive.
“We dosed these animals [with the antisense oligonucleotides] at the age of disease onset and asked what happens, and the answer is we prevented further disease development for the life of those animals with a single dose injection applied at the initial signs of disease,” said Cleveland.
His team initiated clinical trials on this therapy in 2018, just seven years from the date when researchers had first published the data showing the ORF72 mutation caused ALS.
Although it’s an important target for research, inherited ALS accounts for only 10% of the disease’s total cases. In 90% of patients, the condition develops spontaneously due to somatic cell mutations later in life. Many of these cases involve mutations in the TDP-43 gene, which encodes a nuclear protein that regulates Stathmin-2, which in turn plays a critical role in regulating the cytoskeleton in neurons. TDP-43 normally binds the Stathmin-2 precursor RNA and ensures that it gets spliced properly into messenger RNA. Mutations that inactivate TDP-43 cause a loss of functional Stathmin-2, which is a hallmark of sporadic ALS.
Using cultured neurons, Cleveland and his colleagues found that a properly designed antisense oligonucleotide could compensate for the loss of TDP-43 activity, restoring normal RNA splicing and Stathmin-2 expression.
“This now enables a strategy for therapy for sporadic ALS,” said Cleveland.
If the result holds in other preclinical models, he expects to take that approach into clinical trials in 2023.
Besides correcting specific defects within a cell, antisense nucleotides can potentially redirect a cell’s fate entirely. That’s the central theme of another project in Cleveland’s lab, in which the team is causing astrocytes to change their identities. Astrocytes are companion cells in the nervous system that arise from the same stem cells as neurons. Using antisense oligonucleotides, the investigators can suppress two genes that direct cells into the astrocyte lineage, causing them to become neurons instead. Cleveland is initially focusing on treating Parkinson’s disease with this approach, but he explained that “this…conversion of astrocytes into replacement neurons may be broadly applicable for neurogenic disease.”
Huda Zoghbi Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital
Maybe Not So Rare
Huda Zoghbi gave the meeting’s keynote presentation, which covered her work on Rett syndrome. Caused by spontaneous mutations in the MECP2 gene on the X chromosome, Rett syndrome is a progressive neurodegenerative disease that primarily manifests itself in girls. MECP2 is critical for gene regulation in neurons. Because females carry two copies of the X chromosome and an inactivate one in each cell, an inactivating mutation in MECP2 impairs the function of 50% of the affected individual’s neurons. That manifests itself as a rapid regression in motor and cognitive abilities by age two.
In boys, who have only one X chromosome, inactivation of MECP2 is generally lethal before age two. They don’t live long enough to develop the classic symptoms of Rett syndrome. However, recent work has revealed that some males acquire mutations that cause less severe defects in MECP2.
“What we’ve learned is when people carry milder mutations, we will see milder phenotypes, such as mild learning disability with…neuropsychiatric features,” said Zoghbi.
These individuals’ phenotypes can range from autism to hyperactivity or schizophrenia, but most die by middle age due to neurodegeneration. Females with mild defects in MECP2 show non-random inactivation of their X chromosomes, favoring the healthy copy of the gene and enabling them to develop and live normally. Some patients also have duplications in their MECP2 genes, often leading to severe neurological problems and premature death.
To understand the mechanisms driving Rett syndrome, Zoghbi and her colleagues developed a series of genetically modified mice carrying various duplications or mutations in MECP2. Consistent with the findings in humans, these animals display a spectrum of phenotypes depending on the severity of their MECP2 disruptions.
“The brain is very sensitive to the activities and functions of this protein, and we’ve done a lot of studies on both the loss and the gain models,” said Zoghbi.
She and her colleagues found that all types of neurons require functional MECP2 to operate normally.
Mutations affecting different types of neurons can cause a wide range of neurological phenotypes.
Next, Zoghbi and her colleagues tried inactivating MECP2 in excitatory and inhibitory neurons separately. They found that in both cases, animals developed obesity, lost motor coordination, and died young. However, targeting MECP2 only in inhibitory neurons led to more learning and social defects in the animals, while inactivating it only in excitatory neurons caused more anxiety and tremors. These phenotypes represent the downstream effects of the genes MECP2 would normally regulate.
“Given that it’s important for practically every cell, really there’s two major ways you can think of treating this disorder, either gene replacement therapy…or perhaps exploring modulation of the [MECP2 regulatory] circuit,” said Zoghbi.
Taking the latter approach, the investigators implanted electrodes into the brains of mice to deliver small electrical pulses. This type of deep brain stimulation, which has been shown to reverse many types of neuronal signaling and development defects, is already approved for human treatment of several neurological disorders. Stimulating the brains of Rett syndrome model mice leads to significant recovery in their learning, memory, and motor abilities that persists for weeks after treatment.
“It was really quite a dramatic rescue in that all these phenotypes normalize, and their normalization…lasted for several weeks,” said Zoghbi.
The treated animals’ neurons also displayed gene expression patterns similar to wild-type animals, whereas untreated animals showed significant gene dysregulation.
“The Rett brain, at least in mice, is responsive to neuromodulation,” said Zoghbi.
Looking at the MECP2 gene itself, Zoghbi’s team identified regulatory sequences that control its expression level. Altering these sequences to increase or decrease the amount of MECP2 expressed in mice underscored their earlier findings, showing that even modest changes in MECP2 levels led to detectable neurological phenotypes.
Like other speakers at the meeting, Zoghbi and her colleagues are also exploring the potential of antisense oligonucleotides as therapies. That approach seems especially promising for patients with duplications in MECP2 that lead to overexpression of the gene. In mice that recapitulate this condition, the researchers found that treatment with antisense oligonucleotides against MECP2 could reduce the amount of functional protein in neurons down to wild-type levels. The treatment reversed the animals’ motor defects.
Titrating the antisense oligonucleotide dosage also revealed that even modest decreases in excess MECP2 can lead to major improvements in symptoms.
“If you can even partially decrease the protein…you will probably rescue quite a bit of the features of the disease,” Zoghbi said. She added, “I’ve really never worked with a protein that is so exquisitely sensitive to the levels.”
Graciana Diez-Roux, PhD Telethon Institute of Genetics and Medicine
David Fajgenbaum, MD University of Pennsylvania
Anne Heatherington, PhD Takeda Pharmaceuticals
Silver Linings
The meeting’s general session concluded with a panel discussion led by Brad Margus, co-founder and CEO of Cerevance. With a background in business, Margus moved into rare disease drug development after his daughter was diagnosed with ataxia-telangiectasia, a genetic disorder that causes neurodegeneration and immune dysfunction. The panel also featured Graciana Diez-Roux, chief scientific officer at the rare disease-focused Telethon Institute; David Fajgenbaum, a physician-scientist who both studies and suffers from Castleman syndrome; and Anne Heatherington, a data scientist for Takeda Pharmaceuticals with extensive experience studying Duchenne muscular dystrophy.
Panel members discussed the need for improvement in collaborations between patients and researchers.
“There is a lot of miscommunication within the rare disease research space, [but] I think there’s been a really great trend for groups like Takeda and others toward engaging patients in the research process,” said Fajgenbaum, adding that “I also think clinicians can really be a part of this.”
Besides improving clinical trial recruitment, involving patients more directly in research can have far-reaching benefits for scientists.
“It’s incredible how our PhD students, when they have the chance [to interact] with the patients and [get] to know the patients’ organizations…how their motivation and their love for what they do changes,” Diez-Roux said.
Besides increasing collaborations between patients and scientists, all of the panelists endorsed the need for strong, well-defined partnerships with pharmaceutical companies. Margus described his company’s efforts to improve data collection and sharing for ataxia-telangiectasia, which included building a system that uses wearable devices to collect movement data from patients around the clock.
“The data [are] truly owned by the families and the community, and we can make decisions about sharing the data with academics or any researcher in the world in a matter of days,” said Margus.
Good partnerships require more than just good databases, though. Academic researchers accustomed to independent, curiosity-driven experimental design and flexible deadlines sometimes have trouble accommodating pharmaceutical companies’ urgent, goal-directed needs.
“I think the model has to be somewhere between…the industry knowing how to deal with the academic research and academic researchers being open to notice that industries have…different goals in some respects,” said Diez-Roux.
The group also discussed the impact of the COVID-19 pandemic. In the short term, of course, the global shutdown caused by the SARS-CoV-2 virus has halted or delayed many rare disease studies. However, panelists agreed that some of the innovative approaches developed for the pandemic response could transform many aspects of rare disease research in the future.
“We have been very involved in a lot of the COVID alliances, and have been steeped in novel ways of working,” said Takeda’s Heatherington.
As an example, she pointed to the company’s involvement in multi-corporation consortia to develop new therapies and even entirely new platforms for therapies.
“That’s a real breakthrough in terms of how we do our business, that extent of collaboration for [competitors to] come together,” Heatherington continued.
At the same time, “the public is realizing more how important research is, and this goes for COVID, but I think it goes for all diseases,” said Diez-Roux. Both she and Heatherington also pointed out that the pandemic has underscored the potential tradeoffs between speed and safety in therapeutic development, and highlighted the importance of oversight in clinical trials.
The meeting concluded with a virtual poster session, featuring rapid-fire presentations of some of the newest research in rare diseases and offering attendees the ability to interact with the presenters directly. Like the other presentations, the posters represented the diversity and enthusiasm of rare disease researchers.
“What makes me optimistic is the passion and the knowledge…and the fact that we have people that are so dedicated to rare diseases,” said Heatherington.
Lewis C. Cantley’s discovery of the enzyme phosphatidylinositol-3-kinase (PI3K) paved the way for a better understanding of cellular metabolism and its role in human diseases. In response to insulin, PI3K signals through lipids to activate a cellular cascade resulting in increased glucose uptake and subsequent cell growth and division. Cantley’s work has led to new cancer therapies, as PI3K pathway mutations are among the most common to drive cancer development, and a better understanding of insulin resistance in diabetes.
For his groundbreaking discovery of PI3K and critical body of research, Lewis C. Cantley, PhD, of Weill Cornell Medical College, received the 2020 Dr. Paul Janssen Award for Biomedical Research. On September 16, 2020, the New York Academy of Sciences hosted the award symposium to celebrate his achievements. Following Cantley’s award lecture, other experts in the field shared their work on the intersection between cellular metabolism, biology and disease.
Symposium Highlights
Differences in cell metabolism across cancer types may help explain why cancer cells are differentially sensitive to drugs that target metabolism.
The tumor suppressor protein p53 regulates several pathways that manage the production of reactive oxygen species in the mitochondria.
Metabolic pathways represent a powerful and to-date, underappreciated set of therapeutic targets for cancer.
Drugs that regulate metabolic pathways involved in cell differentiation are promising targets for acute myeloid leukemia.
Cancer researchers are coming to appreciate how a patient’s diet can be therapeutically applied and may directly modulate their disease progression and therapeutic response.
Speakers
Lewis C. Cantley, PhD Weill Cornell Medical College
Matthew Vander Heiden, PhD Massachusetts Institute of Technology
Karen Vousden, PhD Francis Crick Institute
Ulrike Philippar, PhD Johnson & Johnson
Costas Lyssiotis, PhD University of Michigan
Discovering Phosphatidylinositol-3-kinase and Its Link to Cancer
Speakers
Lewis C. Cantley Weill Cornell Medical College
Metabolism, Health, and Cancer
Lewis C. Cantley described how the discovery of the enzyme phosphatidylinositol-3-kinase (PI3K) and its signaling pathway led to the development of a new class of cancer drugs called phosphoinositide-3 kinase inhibitors.
He traced his foundational work on PI3K inhibitors to a single slide that his then-graduate student, Malcolm Whitman, produced for a lab meeting in 1987 when he was at Tufts University in Boston. The lab was studying phosphatidylinositol and its role in a signaling cascade regulated to insulin and cell growth. They purified an insulin-activated lipid kinase and found that when activated, it is associated with proteins linked to cancer.
The slide from a 1987 lab meeting that sparked decades of research.
At the time, researchers thought that phosphatidylinositol gets phosphorylated at position 4 on its sugar ring to produce a lipid called PI4P, which is involved in cell regulation. But Whitman found these phosphorylated lipids seemed to have two subtypes—one consistently ran a millimeter further on the gel. “That one-millimeter difference convinced me, as a chemist, that these had to be different species,” Cantley recalled. It turned out that their insulin-regulated kinase—now called PI3 kinase—produces a different molecule, called PI3P, and modulates a previously undiscovered insulin-regulated pathway that regulates the cell’s ability to take up glucose.
Cantley’s team went on to elucidate how PI3K regulates cells’ response to insulin signaling. “Almost every step in the first half of glycolysis is regulated by the PI3 kinase pathway,” he said. Collaborating with a team led by John Blenis, now at Weill Cornell Medicine, Cantley’s also fleshed out the connection between PI3K and multiple other cancer related signaling pathways, such as PTEN, Ras, and mTOR. Cantley called it “really quite remarkable” that “every tumor has at least one of the molecules in this pathway mutated—and many have several.” Indeed, accumulating research since then shows PI3K is one of the most frequently mutated oncogenes in cancer.
Although four different genes encode PI3Ks, just one of them—PIK3CA, which mediates insulin response, is widely mutated in cancers. Yet despite extensive effort, researchers have struggled to get PIK3CA-targeted drugs approved. Because this form of the protein mediates insulin activity, any drug that targets it makes patients insulin-resistant. “So you’re fighting the battle of insulin resistance as you are trying to treat the cancer, Cantley explained.
PIK3CA mutations are especially prevalent in uterine, cervical, breast, and ovarian cancer. In 2009, Cantley received a $12 million grant from the Stand Up To Cancer foundation to form a “dream team” of researchers to investigate how to develop PI3K inhibitors for women’s cancers and explore effective drug combinations. Through this grant, Cantley and his team conducted an early-stage trial of BYL719 (Alpelisib), a PIK3CA inhibitor developed by Novartis, in combination with a chemotherapy agent called letrozole, which lowers estrogen production, in people with metastatic breast cancer. The combination reduced glucose uptake to tumors and extended progression-free survival by roughly one year. The US Food and Drug Administration approved it in 2019.
PI3 Kinase inhibitor BYL719 combined with the chemotherapy agent letrozole reduced glucose uptake from tumors of a patient over the course of one month.
But in that early clinical trial, Cantley noticed that although the drugs’ response correlated with PIK3CA mutations overall, it was uneven; some patients with PIK3CA mutations didn’t respond, and some with mutations in unrelated genes did. “This was not a clear home run,” he said. “I argued, as we looked at the data, that maybe this has something to do with patients’ insulin levels.”
The insulin receptor (IR) is expressed in most tumors. Cantley and his colleagues wanted to interrupt the cycle in which high insulin levels trigger the IR and PI3K activity in the tumor, ultimately driving glucose uptake. To do so, the researchers tracked insulin levels in patients and mice taking PI3 kinase inhibitors. All three drugs they tested raised insulin levels 20-fold. In additional experiments, cells grown from patients with PIK3CA tumors died when exposed to PIK3CA inhibitors but were rescued when insulin was added. “So those ambient levels of insulin really are keeping the tumor alive,” Cantley said.
In subsequent studies, Cantley and his team tested the effects of insulin-lowering drugs, including metformin, as well as a ketogenic diet in mice. They found that a ketogenic diet effectively maintained low glucose and insulin levels while administering a PI3K inhibitor. In mice carrying tumors with a PIK3CA mutation, combining the PI3K inhibitor with a ketogenic diet significantly suppressed tumor growth compared to the control group. “This really tells us that keeping insulin down during the treatment with a PI3 kinase inhibitor could potentially have huge improvements in patient responses,” Cantley said.
Researchers are looking more closely at the effect of a ketogenic diet on the efficacy of PI3K inhibitors for endometrial cancer, breast cancer, and lymphoma. Previously a postdoc in Cantley’s lab and now at Weill Cornell Medicine, Marcus Goncalves is spearheading this effort with a trio of clinical trials currently enrolling patients.
Matthew Vander Heiden Massachusetts Institute of Technology
Karen Vousden Francis Crick Institute
Metabolic Limitations in Cancer
All cells in the body, including cancer cells, exist in different metabolic environments, and thus have different nutritional resources available to them, said Mathew Vander Heiden. Cancer cells have especially high metabolic needs because by definition, they proliferate—doubling the mass of proteins, nucleic acids, and lipids in order to go from one cell to two. Understanding how different types of cancer cells reorganize their metabolic pathways to accomplish this feat can bring insight into the role metabolism plays in cancer therapeutics.
Tissues solve their metabolic needs differently. Metabolism in brain cells, for example, differs from that in liver cells. These differences must be reflected in the gene expression patterns of the tissues’ metabolic networks. When a cell becomes cancerous, it takes its existing metabolic network, based on its environment, and reorganizes it to support its proliferation. That’s why cancers exhibit varied metabolisms and are sensitive to different therapies, Vander Heiden said.
His lab studies how a cell’s environment constrains its metabolic network. In one recent study, the researchers characterized the nutrients available in the interstitial fluid of pancreatic and lung tumors in mice. Transplanting a tumor into a different tissue site changed the nutrients available to it. But the mutational profile of a tumor had a much weaker effect on its metabolism. More recent human data suggests that nutrient profiles in the interstitial fluid of kidney cells and kidney tumor cells is similar. The findings suggest that nutrient availability is an intrinsic property of different tissues, and that cancer must adapt to the food available in a specific area. “That’s a cool idea, because it has a profound effect on how we think about metastasis,” said Vander Heiden. His lab has conducted metabolic phenotyping of mouse cancers transplanted to different tissue sites and found that the most significant outlier in the metabolic profile is in the brain. That matches clinicians’ experience with HER2-positive breast cancer, which tends to respond well to treatment except when the cancer reaches the brain.
To probe tissue-intrinsic metabolic responses, researchers in the lab of Rakesh Jain, also at Harvard, transplanted breast cancer tissue either into mammary fat pads or the brains of mice. They then treated the muse with PI3K inhibitor and found that the tumor shrunk in the mammary tissue but not the brain. Related studies showed that the brain microenvironment partially drove differences in response.
Fatty acid synthesis is increased in the breast cancer tissue transplanted into mouse brain.
In collaboration with Jain’s lab, Vander Heiden’s team implanted tumor cells into the brains and mammary fat pad tissue of mice. They found different metabolic gene expression and activity between the two sites. They also found more glucose uptake into fatty acids in the brain tumor tissue than in the mammary tissue, suggesting that fatty acid synthesis increases in brain metastases. Ultimately, they showed that these differences affect how tumors develop in the brain and facilitate tumor growth. The microenvironment and nutritional differences of tumor cells is key to understanding varied responses to metabolism-targeted therapies.
Metabolic Control of Tumor Progression
P53 is a key tumor suppressor gene. Some seven million cancer patients per year carry a mutant version of it. However, virtually all tumors, even those with a normal p53 gene, have lost p53’s tumor suppressive activity, said Karen Vousden. Early research showed that p53 suppresses tumors but activates cell death and senescence, killing tumor cells. But more recently, Vousden and others report another type of p53 activity, in which it contributes to the cell’s survival and its ability to adapt to stressful conditions, such as the accumulation of reactive oxygen species (ROS). Her lab is examining the gene’s role in metabolic signals such as nutrient fluctuation and oxidative stress.
Next, they looked at p53’s role in adapting to nutrient starvation. Comparing cell lines with p53 to without it, they found that p53 mitigates oxidative stress caused by cells lacking amino acids the body normally produces, such as serine. The absence of serine had stronger negative effects in cells without p53, reflecting the gene’s ability to drive antioxidant defense, Vousden said.
To look at how p53 protects cells from oxidation, the researchers focused on a downstream enzyme called TIGAR. Although the protein’s full suite of functions is still unknown, it promotes the production of NADPH, a molecule that supports antioxidant activity. Cells that lack TIGAR are more sensitive to oxidative stress and can’t maintain NADPH levels. Studies suggest that this effect is specific to mitochondria. Organoids lacking TIGAR have a much higher level of toxic ROS than organoids expressing the enzyme, and treating the cells with mitochondrial antioxidants — but not membrane antioxidant — mitigates this effect.
p53 controls reactive oxygen species via downstream molecules, including TIGAR.
Vousden’s lab also studied the effects of TIGAR during various stages of tumor development. They found that loss of TIGAR impedes tumor growth in both the pancreatic and intestinal adenoma models. Interestingly, mice with these tumors also had an increased rate of lung metastasis. Treating these mice with antioxidants mitigated the effect on lung metastasis, suggesting that ROS influences cancer development differently at different stages of the disease.
Targeting Metabolism and Differentiation Therapy in AML
Acute Myeloid Leukemia (AML) is a critical disease area in Janssen’s drug discovery effort. A clinically heterogenous disease with multiple subtypes, AML is characterized by a diverse landscape of chromosomal abnormalities and gene mutations. One hallmark of AML, myeloid differentiation block, is linked to metabolism, said Ulrike Philippar. Myeloid differentiation in normal cells results in the generation of mature blood cells from hematopoietic stem cells. This process is blocked in AML cells, resulting in uncontrolled proliferation of progenitors, and ultimately, aggressive leukemia. Differentiation therapies have thus been a recent focus of drug development for AML, and unmet medical need for the disease remains high.
Philippar first described two US Food and Drug Administration-approved differentiation therapies. The first, called all-trans retinoic acid, or ATRA, was first used in 1985 to treat acute promyelocytic leukemia (APL), a subtype of AML. ATRA binds to a translocated protein in APL, leading to disruption of the differentiation block. The second therapy, IDH1 and IDH2 inhibitors, treats AML patients with an IDH mutation. The drug came to be used in combination with arsenic trioxide. These agents bind to a translocated protein in APL, disrupting the differentiation block.
Philippar and her team conducted two CRISPR/Cas9 screens to identify novel targets that inhibit proliferation in cell lines in vitro and/or in a bone marrow transplant mouse model in vivo. They identified dihydroorotate dehydrogenase (DHODH), an enzyme located in the mitochondrial membrane involved in pyrimidine synthesis. DHODH is also essential for the production of the nucleotide uridine monophosphate (UMP), the first building block in the production of RNA and DNA synthesis. Researchers, including Philippar and her colleagues, have shown that malignant cells, particularly AML cells, are metabolically dependent on pyrimidine production because they cannot salvage sufficient uridine from the extracellular environment.
Janssen identified a potent, selective proprietary DHODH inhibitor. The molecule strongly inhibited proliferation in 25 AML cancer cell lines. Janssen’s DHODH inhibitor also showed strong antiproliferative activity in primary AML samples. A few patient samples appeared to be resistant to the drug, and Janssen researchers will continue to investigate the basis of that resistance.
Preclinical studies suggest that DHODH inhibition is an effective treatment strategy for AML
The researchers also probed the DHODH inhibitor’s mechanism of action. The molecule induced cell cycle arrest in S phase, resulting in increased apoptosis. Furthermore, treatment with the DHODH inhibitor also increased cellular differentiation in AML cell lines. Adding excess uridine can reverse these effects, which validates the inhibitor’s on-target activity. Janssen’s DHODH inhibitor led to tumor regression in subcutaneous AML xenografts and increased the lifespan of mice bearing disseminated AML cancer cells approximately twofold.
Philippar said that DHODH inhibitor therapy takes away the pyrimidine source and leads to starvation of cells by forcing them into a fasting state. The researchers hypothesize that normal cells can tolerate fasting and recover; however, leukemia and other cancer cells cannot tolerate fasting because they require higher pyrimidine synthesis. Hence, prolonged pyrimidine starvation of leukemia cells results in a cell fate decision leading to differentiation, and ultimately, apoptosis and tumor regression.
DHODH confirms the relevance of metabolic targets for AML. However, the clinical diversity of AML may be a challenge for metabolic drugs, and researchers will have to better understand patient selection as they develop these drugs in the clinic.
Lewis C. Cantley, PhD Weill Cornell Medical College
Matthew Vander Heiden, PhD Massachusetts Institute of Technology
Karen Vousden, PhD Francis Crick Institute
Ulrike Philippar, PhD Johnson & Johnson
Panelists reflected on what moderator Costas Lyssiotis, a former postdoctoral student in Cantley’s laboratory, framed as a key theme of the symposium: harnessing diet as a way to treat cancer.
Cantley stated that previous efforts to use diet to understand cancer have largely failed, because getting trial participants to adhere to a diet and accurately report what they eat is nearly impossible. In a recent clinical trial that tested a ketogenic diet as a neoadjuvant for endometrial cancer, Cantley’s collaborator Marcus Goncalves used the metabolic kitchen at Weill Cornell Medical School to generate 21 meals per week for each trial participant. Weekly blood draws made it possible for researchers to be sure patients adhered to the diet. “To show [that] a dietary intervention works, you have to do it in a very controlled manner, not just tell people what they should eat and hope that they find it at the market,” Cantley said. Ultimately, the aim is to get dietary interventions approved as therapies so that insurance companies will cover them. He noted that companies such as Bayer and Novartis are excited by the possibility because it can potentiate pharmaceutical therapies. “We should hold diets to the same standards we hold drugs to,” said Cantley.
Vander Heiden agreed about the need for more rigorous dietary trials. His lab found that alterations in diet led to changes in the composition of metabolic molecules in interstitial fluid, and presumably those changes extend to nutrients available in different tissues and in blood. Animal studies are especially useful for identifying how nutrient availability changes with diet, he said; and they might point to unexpected differences beyond those already identified for the glucose and insulin pathway and serine.
Translating dietary interventions from animals to humans may not be straightforward. “What happens in mice doesn’t necessarily happen in people,” Vousden said. She and her colleagues are developing a serine-depleted diet for people with cancer, along the lines of low phenylalanine diets prescribed for children with phenylketonuria, an inherited condition that raises the level of the amino acid building block phenylalanine in the blood.
Any nutrient may be modulated for cancer therapy, but researchers must have a firm mechanistic understanding of why that alteration might work. “I think that’s at the heart of whether it will be successful, and whether people will follow these diets,” Vousden said. A solid grounding in the mechanism underlying a possible dietary intervention’s effect can help motivate patients to follow a restrictive diet for their clinical benefit.
Philippar noted that so far, considering diet is not standard practice in industry drug development. But it is potentially important, not just for targets that modulate the metabolism but also for immunological approaches. Researchers now know that diet can affect gut bacteria, which might in turn affect drug response. In clinical trials run by Janssen, she explained, researchers generally check whether a patient is in a fed or fasted state when receiving an experimental therapy, but they do not check on what exactly the patient has eaten.
Single-cell RNA sequencing makes it possible to look at the tumor microenvironment and to determine how changes in diet affect the expression levels of immune genes. In 2019, Cantley, Goncalves and others showed that just one, 12-ounce serving of a sugary drink such as orange juice, apple juice, or soda each day enhances intestinal tumors in mice. A combination of fructose and glucose causes the effect, the researchers found, and knocking out a molecule called ketohexakinase, which phosphorylates fructose, limits tumor growth. The connection to sugary drinks “is very surprising,” says Cantley, “but I think it may explain why we’ve almost tripped the rate of early onset colorectal cancer in young adults in the last 20 years.”
Artificial intelligence is quickly becoming a ubiquitous part of our daily lives. What can we expect as this technology continues to grow? And how will it impact you?
Published September 14, 2020
By Liang Dong
Alexandra Boltasseva, PhD
From virtual assistants like Siri to self-driving cars and computer-aided medical diagnoses, artificial intelligence (AI) affects our lives with unprecedented speed. Slowly but steadily, scientists in a broad range of fields have started to embrace AI in their research, hoping to significantly reduce the time needed to achieve new discoveries. This trend has become more obvious in the physical sciences, and in the field of materials science in particular, which is focused on the discovery and production of new, advanced materials imbued with desirable properties or functions. Think: screens of foldable smartphones; batteries that power electric cars; or materials that bend light around them, rendering them invisible.
How exactly could AI help materials scientists? We recently interviewed three honorees of the Blavatnik Awards for Young Scientists, Alexandra Boltasseva, PhD, Professor of Electrical and Computer Engineering at Purdue University; Léon Bottou, PhD, Principal Researcher at Facebook AI Research; and Sergei V. Kalinin, PhD, Corporate Fellow at Oak Ridge National Laboratory, who are contributing to an upcoming virtual symposium on October 6 and 7, AI for Materials: From Discovery to Production. Here’s what they had to say about the opportunities, as well as the challenges, in this rising field.
It is only recently that researchers in the physical sciences, like materials scientists, have begun to incorporate AI techniques into their work. Why do we need to take advantage of AI for this field? What benefits may AI offer materials science?
Kalinin
Sergei V. Kalinin, PhD
AI offers a set of powerful tools to explore large volumes of multidimensional data in the physical sciences, and promises to uncover hidden functional relationships between the physical properties that we can observe. As such, AI methods are poised to become an inseparable part of all physical sciences, to enable discovery and hypothesis-driven research and to guide planning of experiments. We can take advantage of a broad range of AI techniques—from multivariate statistics to convolutional networks, unsupervised and semi-supervised methods, Gaussian processing, and reinforcement learning.
In addition, the proliferation of laboratory automation in areas from materials synthesis to imaging of materials’ molecular structures opens up broad opportunities for AI-driven experiments. For example, we will be able to adopt large-scale robotic systems or the microscale lab-on-a-chip platforms in our experiments, producing thousands or more materials in a single process.
Boltasseva
My own field, photonics, has truly been transformed by the concept of “inverse design,” meaning scientists input desired performances of photonic systems into computers and run physics-informed algorithms to figure out the best possible optical designs. The daunting challenge of this field lies in the inconceivably high computational power required for an exhaustive search within the extremely large, hyper-dimensional space of optical design parameters and constituent materials. Merging AI techniques with photonics is expected to not only enhance and enrich the design space, but, most importantly, to unlock novel functionalities and bring about disruptive performance improvements.
As compared to life sciences and pharmaceutical sciences, the application of AI in physical sciences is at least 10 years behind. What do you think is the biggest challenge for applying AI in physical sciences? How could the AI and physical sciences communities work together to address these challenges?
Bottou
Léon Bottou, PhD
Using machine learning in physical sciences is not an obvious proposition. Recent advances in AI have shown how tasks in computer science, such as computer vision and machine translation, can be achieved using big data. Yet it would be unwise to claim that this success can be replicated in all scientific fields. Big data only reveal statistical correlations that are not always indicative of the causal relations that physicists often seek. To solve this question, the AI and physics communities may take the strategy of defining a hierarchy of problems for which one could envision using AI, such as:
Visualizing or measuring an ongoing physical phenomenon. These problems are the most accessible to AI/machine learning because they can directly leverage recent advances in computer vision and signal analysis in collecting data from physical experiments and computations.
Explaining a physical phenomenon. These problems belong to the next rung of difficulty because we need AI/machine learning systems that incorporate enough of our current knowledge of physics, and can then clarify the phenomenon of interest by constructing something interpretable on top of our current knowledge.
Designing a physical system that leverages a certain phenomenon in new ways. These are by far the most difficult problems, because they require AI/machine learning systems to accurately predict how the physical phenomenon will be affected by changes that are not included or prominent in the experimental data on which AI models have been trained.
Boltasseva
The physical sciences community should ultimately build extensive databases to unleash the power of AI. We should even set up an ‘optical structures and materials genome’ project to construct a comprehensive dataset of photonic concepts, architectures, components, and photonic materials to enable hierarchical machine learning algorithms that could provide ultimate-efficiency devices.
Kalinin
I agree with Alexandra. AI tends to proliferate in the communities that adopt the model of open sharing of codes and data. While some areas of physics research have undergone this transformation, many more require both enabling tools and proof-of-benefit to accelerate this process.
I also want to add on to Léon’s comment on the fundamental difference between the AI and physics communities. AI starts with purely correlative models, and tends to rely on big data. In comparison, research in physical sciences is strongly based on prior knowledge to explore the cause and effect relationships, and often assumes the presence of simple rules or descriptors that can give rise to complex behaviors in macroscopic systems. Experiments in physical sciences can give rise to huge data volumes, but these data can pertain only to one specific situation of the system and hence are not “big.”
In order to further leverage the benefits of AI in physical sciences, researchers have to possess both sufficient domain knowledge in physical sciences and expertise in machine learning, or forge robust interdisciplinary collaborations. Conferences like AI for Materials will help researchers in both fields form these kinds of interdisciplinary teams.



















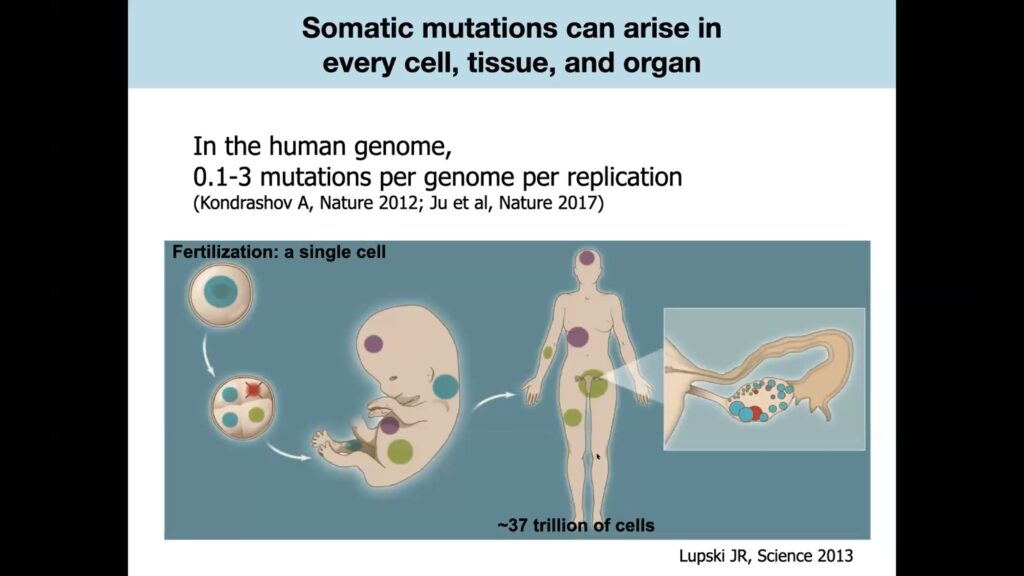


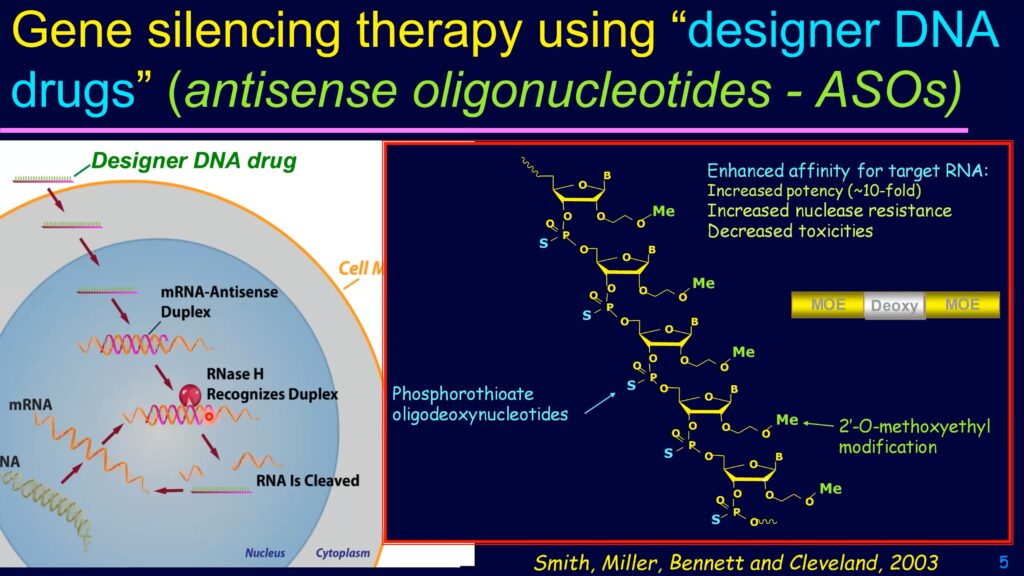

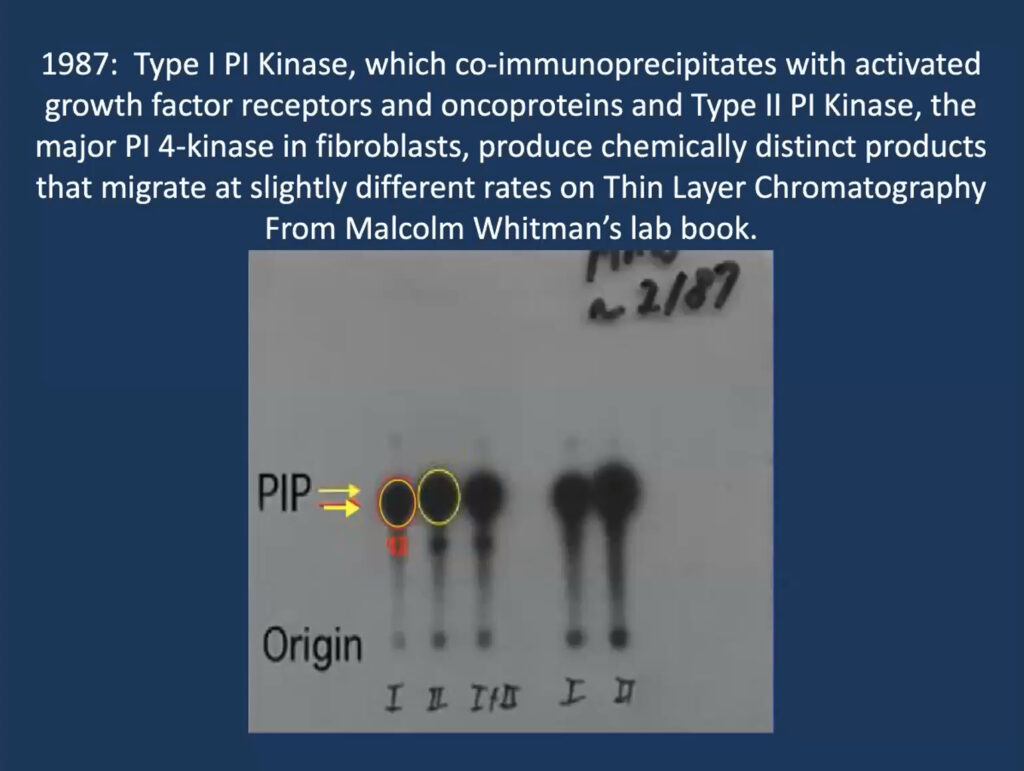
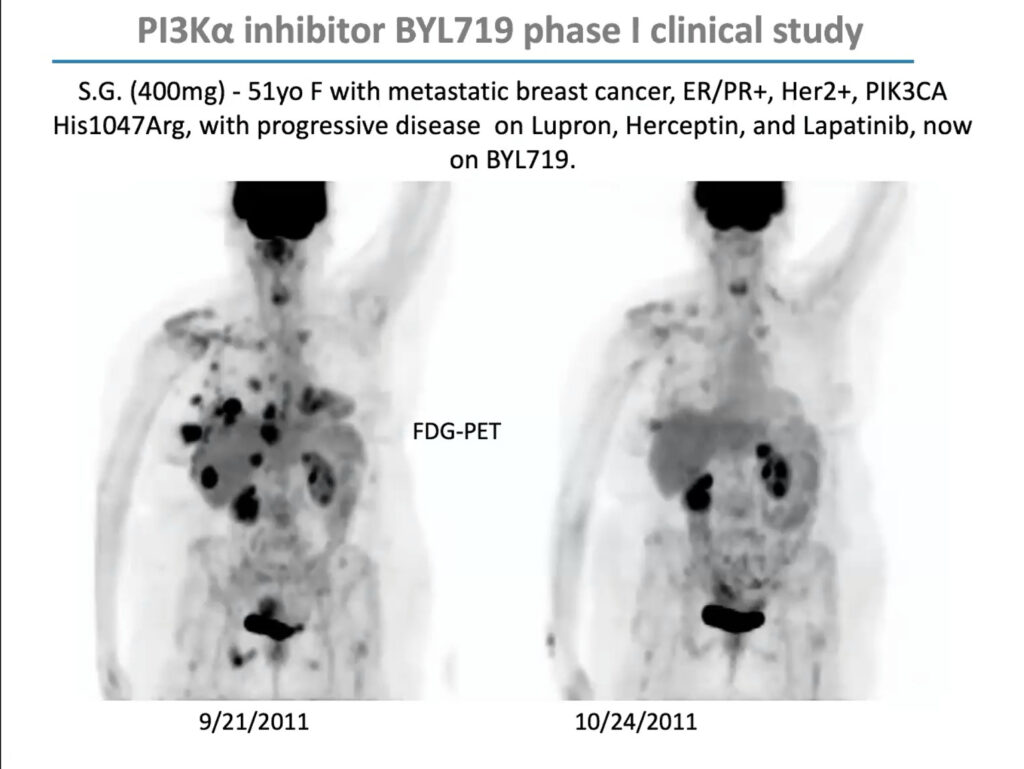
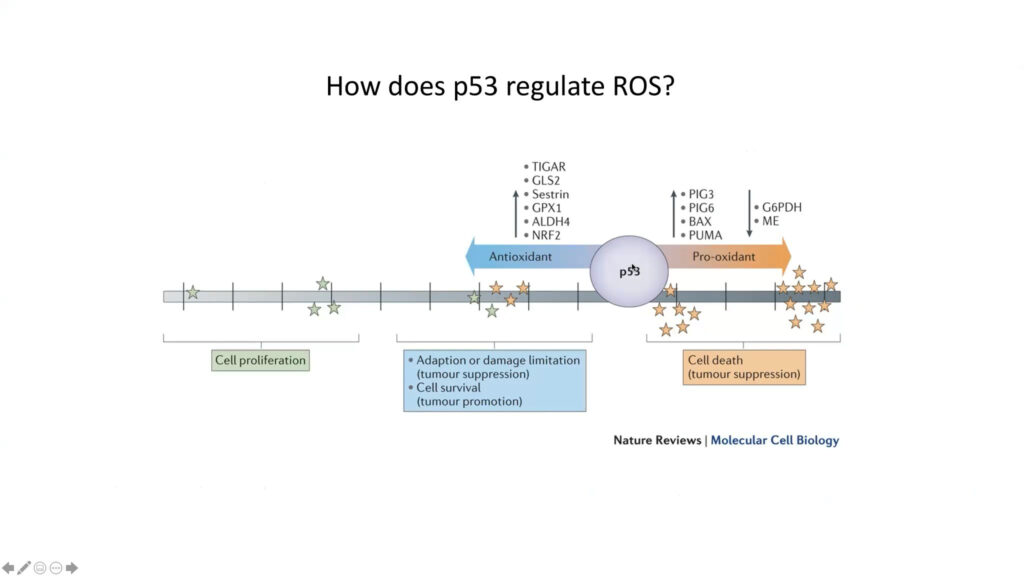
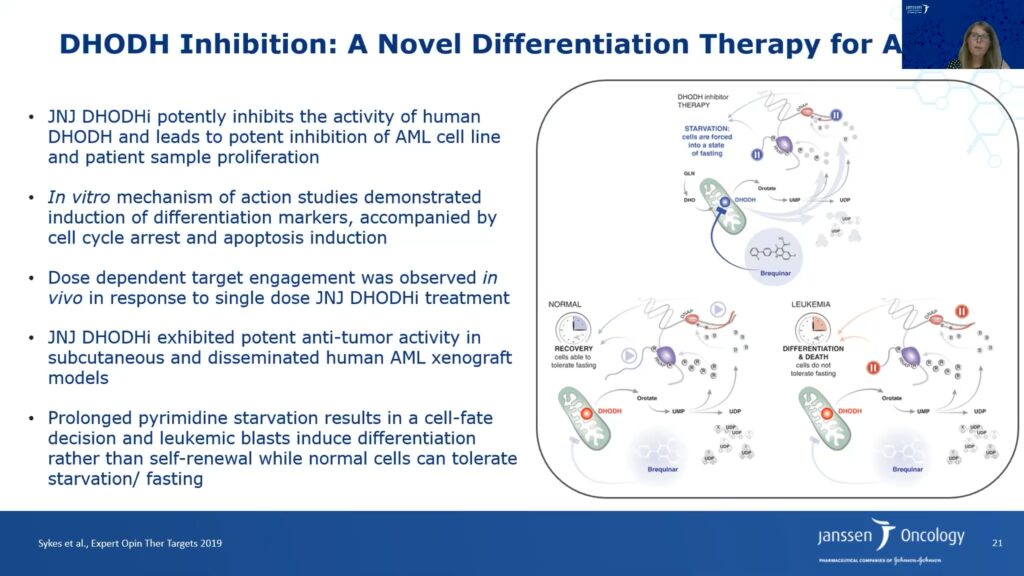




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}